Fabian Kolesch, Marco Sohn, Andreas Rempel, Pia Hippel, Roland Wittler
{"title":"SANS ambages: phylogenomics with abundance-filter, multi-threading, and bootstrapping on amino-acid or genomic sequences.","authors":"Fabian Kolesch, Marco Sohn, Andreas Rempel, Pia Hippel, Roland Wittler","doi":"10.1186/s12859-025-06204-2","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>The increasing amount of available genome sequence data enables large-scale comparative studies. A common task is the inference of phylogenies- a challenging task if close reference sequences are not available, genome sequences are incompletely assembled, or the high number of genomes precludes multiple sequence alignment in reasonable time. SANS is an alignment-free, whole-genome based approach for phylogeny estimation.</p><p><strong>Results: </strong>Here we present a new implementation SANS ambages with a significantly increased application spectrum. It offers additional types of input data, parallelized processing, and bootstrapping. The source code (C++), documentation, and example data are freely available for download at: https://github.com/gi-bielefeld/sans . SANS can also be launched via the web-interface of the CloWM platform- free of charge, with a standard Life Science account: https://clowm.bi.denbi.de/workflows/0194b78f-9696-7402-a2b8-858508733618/ .</p><p><strong>Conclusions: </strong>The new version not only shortens processing time on large datasets immensely by parallelization. Being able to also process amino acid sequences and offering a filter for low-abundant DNA read segments also enables new application cases. Bootstrapping and integrated visualization ease and enrich the interpretation of the resulting phylogenies.</p>","PeriodicalId":8958,"journal":{"name":"BMC Bioinformatics","volume":"26 1","pages":"227"},"PeriodicalIF":3.3000,"publicationDate":"2025-09-02","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12403963/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"BMC Bioinformatics","FirstCategoryId":"99","ListUrlMain":"https://doi.org/10.1186/s12859-025-06204-2","RegionNum":3,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"BIOCHEMICAL RESEARCH METHODS","Score":null,"Total":0}
引用次数: 0
Abstract
Background: The increasing amount of available genome sequence data enables large-scale comparative studies. A common task is the inference of phylogenies- a challenging task if close reference sequences are not available, genome sequences are incompletely assembled, or the high number of genomes precludes multiple sequence alignment in reasonable time. SANS is an alignment-free, whole-genome based approach for phylogeny estimation.
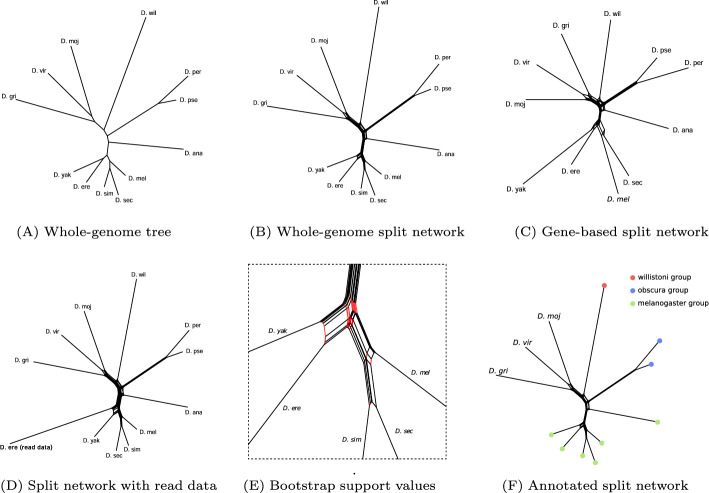
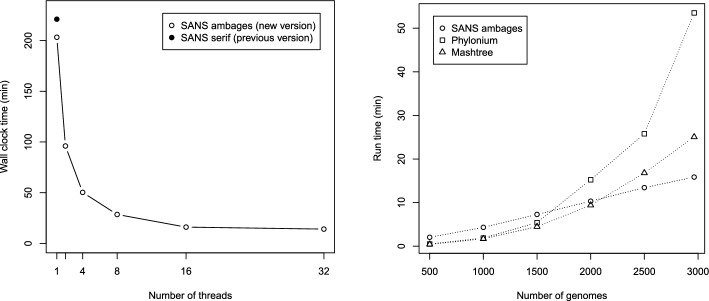
Results: Here we present a new implementation SANS ambages with a significantly increased application spectrum. It offers additional types of input data, parallelized processing, and bootstrapping. The source code (C++), documentation, and example data are freely available for download at: https://github.com/gi-bielefeld/sans . SANS can also be launched via the web-interface of the CloWM platform- free of charge, with a standard Life Science account: https://clowm.bi.denbi.de/workflows/0194b78f-9696-7402-a2b8-858508733618/ .
Conclusions: The new version not only shortens processing time on large datasets immensely by parallelization. Being able to also process amino acid sequences and offering a filter for low-abundant DNA read segments also enables new application cases. Bootstrapping and integrated visualization ease and enrich the interpretation of the resulting phylogenies.
期刊介绍:
BMC Bioinformatics is an open access, peer-reviewed journal that considers articles on all aspects of the development, testing and novel application of computational and statistical methods for the modeling and analysis of all kinds of biological data, as well as other areas of computational biology.
BMC Bioinformatics is part of the BMC series which publishes subject-specific journals focused on the needs of individual research communities across all areas of biology and medicine. We offer an efficient, fair and friendly peer review service, and are committed to publishing all sound science, provided that there is some advance in knowledge presented by the work.


 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


