{"title":"SV-MeCa: an XGBoost-based meta-caller approach for structural variant calling from short-read data.","authors":"Rudel Christian Nkouamedjo Fankep, Arda Söylev, Anna-Lena Kobiela, Jochen Blom, Corinna Ernst, Susanne Motameny","doi":"10.1186/s12859-025-06246-6","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>Calling structural variants (SVs), i.e., genomic alterations of ≥50bp, from whole genome short-read data remains challenging, as existing callers are known to lack accuracy and robustness. Therefore, meta-caller approaches combining the results of multiple standalone tools in a consensus set of reported SV calls, are widely used. Here, SV-MeCa (Structural Variant Meta-Caller) is presented, the first SV meta-caller incorporating variant-specific quality metrics from individual VCF outputs, rather than relying solely on number and combination of tools supporting consensus SV calls. In addition, SV-MeCa offers a suitable score to rank obtained consensus SV calls according to evidence of representing true positive calls, i.e., real-world variants.</p><p><strong>Results: </strong>SV-MeCa applies seven standalone SV callers and merges resulting deletion and insertion calls into a union VCF file using SURVIVOR. For each entry in the SURVIVOR-generated consensus, caller-specific quality measures are extracted from corresponding standalone VCF files, and serve as input for an either deletion- or insertion-specific XGBoost decision tree classifier, which was previously trained on the HG002 SV benchmark data provided by the Genome in a Bottle consortium. The SV-MeCa XGBoost models assign a probability to (consensus) SV calls to represent true positive calls, which can be used for ranking the final output according to evidence. Performance of SV-MeCa and four previously published meta-caller approaches were evaluated based on autosomal SV calls in samples curated by the Human Genome Structural Variation Consortium, Phase 2. With regard to F[Formula: see text] scores, which were 0.58 on average for deletions and 0.42 on average for insertions, SV-MeCa outperformed the other meta-callers. With regard to precision, only ConsensuSV achieved higher values (0.97 versus 0.64 on average for deletions, 0.75 versus 0.53 on average for insertions), and with regard to recall, SV-MeCa was outperformed exclusively by Meta-SV for deletions (0.55 versus 0.53).</p><p><strong>Conclusions: </strong>SV-MeCa, publicly available at https://github.com/ccfboc-bioinformatics/SV-MeCa , outperforms existing SV meta-caller approaches by taking variant-specific quality measures into account. Moreover, due to the XGBoost prediction probabilities serving as scores, the output of SV-MeCa can be continuously adjusted to user needs in terms of sensitivity and precision.</p>","PeriodicalId":8958,"journal":{"name":"BMC Bioinformatics","volume":"26 1","pages":"218"},"PeriodicalIF":3.3000,"publicationDate":"2025-08-20","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12366149/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"BMC Bioinformatics","FirstCategoryId":"99","ListUrlMain":"https://doi.org/10.1186/s12859-025-06246-6","RegionNum":3,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"BIOCHEMICAL RESEARCH METHODS","Score":null,"Total":0}
引用次数: 0
Abstract
Background: Calling structural variants (SVs), i.e., genomic alterations of ≥50bp, from whole genome short-read data remains challenging, as existing callers are known to lack accuracy and robustness. Therefore, meta-caller approaches combining the results of multiple standalone tools in a consensus set of reported SV calls, are widely used. Here, SV-MeCa (Structural Variant Meta-Caller) is presented, the first SV meta-caller incorporating variant-specific quality metrics from individual VCF outputs, rather than relying solely on number and combination of tools supporting consensus SV calls. In addition, SV-MeCa offers a suitable score to rank obtained consensus SV calls according to evidence of representing true positive calls, i.e., real-world variants.
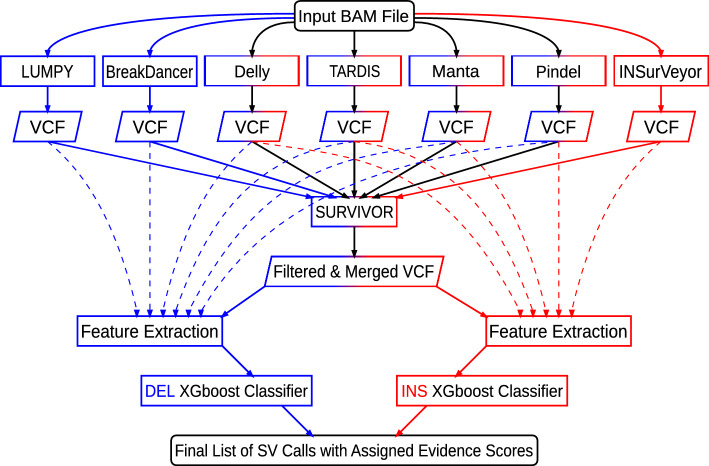
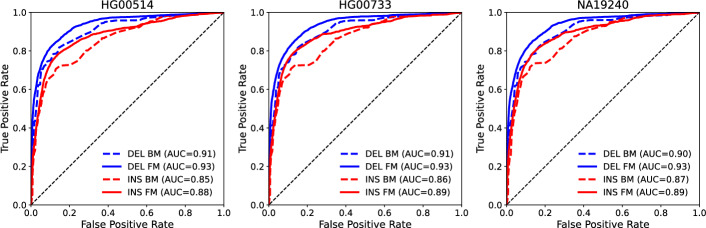
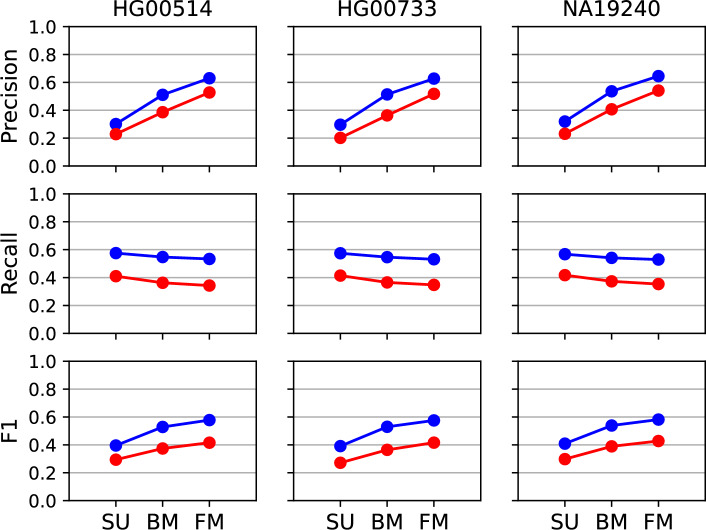
Results: SV-MeCa applies seven standalone SV callers and merges resulting deletion and insertion calls into a union VCF file using SURVIVOR. For each entry in the SURVIVOR-generated consensus, caller-specific quality measures are extracted from corresponding standalone VCF files, and serve as input for an either deletion- or insertion-specific XGBoost decision tree classifier, which was previously trained on the HG002 SV benchmark data provided by the Genome in a Bottle consortium. The SV-MeCa XGBoost models assign a probability to (consensus) SV calls to represent true positive calls, which can be used for ranking the final output according to evidence. Performance of SV-MeCa and four previously published meta-caller approaches were evaluated based on autosomal SV calls in samples curated by the Human Genome Structural Variation Consortium, Phase 2. With regard to F[Formula: see text] scores, which were 0.58 on average for deletions and 0.42 on average for insertions, SV-MeCa outperformed the other meta-callers. With regard to precision, only ConsensuSV achieved higher values (0.97 versus 0.64 on average for deletions, 0.75 versus 0.53 on average for insertions), and with regard to recall, SV-MeCa was outperformed exclusively by Meta-SV for deletions (0.55 versus 0.53).
Conclusions: SV-MeCa, publicly available at https://github.com/ccfboc-bioinformatics/SV-MeCa , outperforms existing SV meta-caller approaches by taking variant-specific quality measures into account. Moreover, due to the XGBoost prediction probabilities serving as scores, the output of SV-MeCa can be continuously adjusted to user needs in terms of sensitivity and precision.
期刊介绍:
BMC Bioinformatics is an open access, peer-reviewed journal that considers articles on all aspects of the development, testing and novel application of computational and statistical methods for the modeling and analysis of all kinds of biological data, as well as other areas of computational biology.
BMC Bioinformatics is part of the BMC series which publishes subject-specific journals focused on the needs of individual research communities across all areas of biology and medicine. We offer an efficient, fair and friendly peer review service, and are committed to publishing all sound science, provided that there is some advance in knowledge presented by the work.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


