Océane Fourquet, Martin S Krejca, Carola Doerr, Benno Schwikowski
{"title":"Towards the genome-scale discovery of bivariate monotonic classifiers.","authors":"Océane Fourquet, Martin S Krejca, Carola Doerr, Benno Schwikowski","doi":"10.1186/s12859-025-06253-7","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>Bivariate monotonic classifiers (BMCs) are based on pairs of input features. Like many other models used for machine learning, they can capture nonlinear patterns in high-dimensional data. At the same time, they are simple and easy to interpret. Until now, the use of BMCs on a genome scale was hampered by the high computational complexity of the search for pairs of features with a high leave-one-out performance estimate.</p><p><strong>Results: </strong>We introduce the fastBMC algorithm, which drastically speeds up the identification of BMCs. The algorithm is based on a mathematical bound for the BMC performance estimate while maintaining optimality. We show empirically that fastBMC speeds up the computation by a factor of at least 15 already for a small number of features, compared to the traditional approach. For two of the three smaller biomedical datasets that we consider here, the resulting possibility of considering much larger sets of features translates into significantly improved classification performance. As an example of the high degree of interpretability of BMCs, we discuss a straightforward interpretation of a BMC glioblastoma survival predictor, an immediate novel biomedical hypothesis, options for biomedical validation, and treatment implications. In addition, we study the performance of fastBMC on a larger and well-known breast cancer dataset, validating the benefits of the BMCs for biomarker identification and biomedical hypothesis generation.</p><p><strong>Conclusion: </strong>fastBMC enables the rapid construction of robust and interpretable ensemble models using BMC, facilitating the discovery of gene pairs predictive of relevant phenotypes and their interaction in that context.</p><p><strong>Availability: </strong>We provide the first open-source implementation for learning BMCs, a Python implementation of fastBMC in particular, and Python code to reproduce the fastBMC results on real and simulated data in this paper, at https://github.com/oceanefrqt/fastBMC .</p>","PeriodicalId":8958,"journal":{"name":"BMC Bioinformatics","volume":"26 1","pages":"228"},"PeriodicalIF":3.3000,"publicationDate":"2025-09-02","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12403431/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"BMC Bioinformatics","FirstCategoryId":"99","ListUrlMain":"https://doi.org/10.1186/s12859-025-06253-7","RegionNum":3,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"BIOCHEMICAL RESEARCH METHODS","Score":null,"Total":0}
引用次数: 0
Abstract
Background: Bivariate monotonic classifiers (BMCs) are based on pairs of input features. Like many other models used for machine learning, they can capture nonlinear patterns in high-dimensional data. At the same time, they are simple and easy to interpret. Until now, the use of BMCs on a genome scale was hampered by the high computational complexity of the search for pairs of features with a high leave-one-out performance estimate.
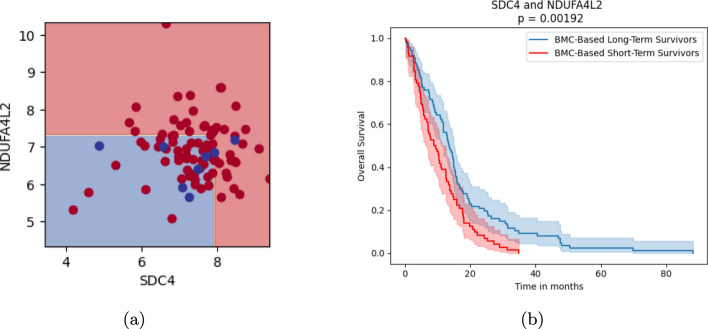
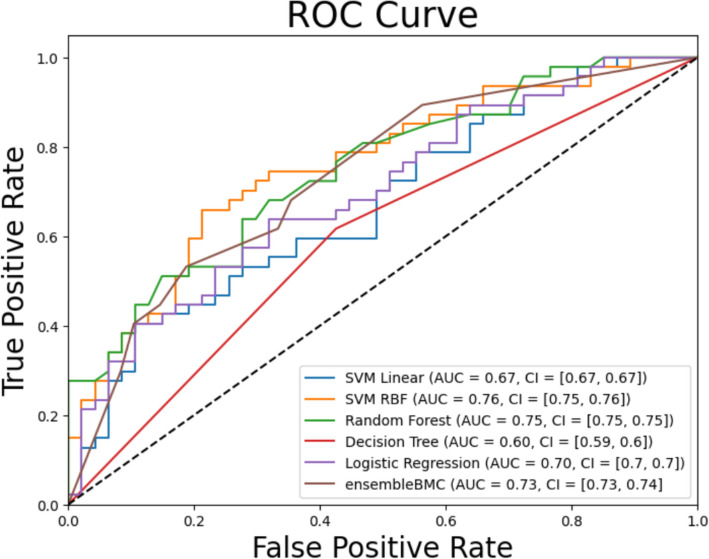
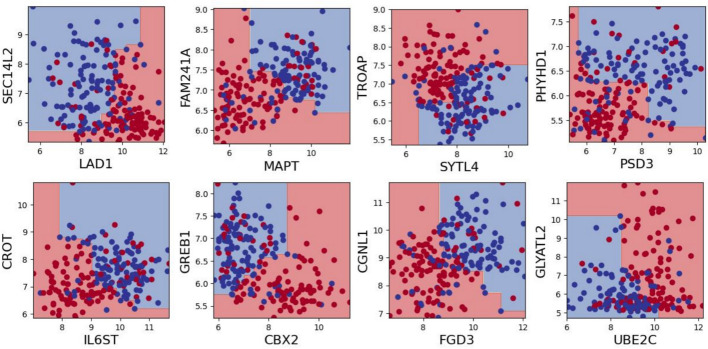
Results: We introduce the fastBMC algorithm, which drastically speeds up the identification of BMCs. The algorithm is based on a mathematical bound for the BMC performance estimate while maintaining optimality. We show empirically that fastBMC speeds up the computation by a factor of at least 15 already for a small number of features, compared to the traditional approach. For two of the three smaller biomedical datasets that we consider here, the resulting possibility of considering much larger sets of features translates into significantly improved classification performance. As an example of the high degree of interpretability of BMCs, we discuss a straightforward interpretation of a BMC glioblastoma survival predictor, an immediate novel biomedical hypothesis, options for biomedical validation, and treatment implications. In addition, we study the performance of fastBMC on a larger and well-known breast cancer dataset, validating the benefits of the BMCs for biomarker identification and biomedical hypothesis generation.
Conclusion: fastBMC enables the rapid construction of robust and interpretable ensemble models using BMC, facilitating the discovery of gene pairs predictive of relevant phenotypes and their interaction in that context.
Availability: We provide the first open-source implementation for learning BMCs, a Python implementation of fastBMC in particular, and Python code to reproduce the fastBMC results on real and simulated data in this paper, at https://github.com/oceanefrqt/fastBMC .
期刊介绍:
BMC Bioinformatics is an open access, peer-reviewed journal that considers articles on all aspects of the development, testing and novel application of computational and statistical methods for the modeling and analysis of all kinds of biological data, as well as other areas of computational biology.
BMC Bioinformatics is part of the BMC series which publishes subject-specific journals focused on the needs of individual research communities across all areas of biology and medicine. We offer an efficient, fair and friendly peer review service, and are committed to publishing all sound science, provided that there is some advance in knowledge presented by the work.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


