{"title":"From AI-Driven Sequence Generation to Molecular Simulation: A Comprehensive Framework for Antimicrobial Peptide Discovery","authors":"Chunsuo Tian, , , Yuelei Hao, , , Haohao Fu*, , , Xueguang Shao*, , and , Wensheng Cai*, ","doi":"10.1021/acs.jcim.5c00892","DOIUrl":null,"url":null,"abstract":"<p >Antimicrobial Peptides (AMPs) are a promising strategy to address bacterial resistance, yet only a limited number have advanced to clinical trials. Recent advances in deep learning provide new opportunities for AMP design. Here, we propose an integrated computational framework combining deep learning with molecular simulation to systematically design and screen novel AMPs. Employing a naïve character-string-based generative adversarial network (GAN), we generated 50 candidate sequences, which were preliminarily screened by the antibacterial peptide discriminative network PGAT-ABPp along with key physicochemical parameters. This screening identified 9 potential functional AMPs. Subsequent molecular dynamics simulations revealed that two peptides can induce water pore formation in bacterial membranes within a limited simulation period, suggesting their potential antibacterial activity. These two peptides were synthesized and tested in vitro, demonstrating efficacy against both Gram-negative (<i>E. coli</i>) and Gram-positive (<i>S. aureus</i>) bacteria, thus confirming their clinical potential. This study not only discovered two novel AMPs but also established a cost-effective design strategy, highlighting the broad applicability of this approach for AMP discovery.</p>","PeriodicalId":44,"journal":{"name":"Journal of Chemical Information and Modeling ","volume":"65 18","pages":"9566–9575"},"PeriodicalIF":5.3000,"publicationDate":"2025-08-29","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Chemical Information and Modeling ","FirstCategoryId":"92","ListUrlMain":"https://pubs.acs.org/doi/10.1021/acs.jcim.5c00892","RegionNum":2,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"CHEMISTRY, MEDICINAL","Score":null,"Total":0}
引用次数: 0
Abstract
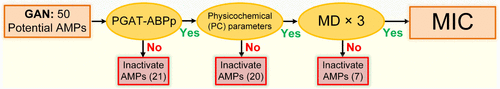
Antimicrobial Peptides (AMPs) are a promising strategy to address bacterial resistance, yet only a limited number have advanced to clinical trials. Recent advances in deep learning provide new opportunities for AMP design. Here, we propose an integrated computational framework combining deep learning with molecular simulation to systematically design and screen novel AMPs. Employing a naïve character-string-based generative adversarial network (GAN), we generated 50 candidate sequences, which were preliminarily screened by the antibacterial peptide discriminative network PGAT-ABPp along with key physicochemical parameters. This screening identified 9 potential functional AMPs. Subsequent molecular dynamics simulations revealed that two peptides can induce water pore formation in bacterial membranes within a limited simulation period, suggesting their potential antibacterial activity. These two peptides were synthesized and tested in vitro, demonstrating efficacy against both Gram-negative (E. coli) and Gram-positive (S. aureus) bacteria, thus confirming their clinical potential. This study not only discovered two novel AMPs but also established a cost-effective design strategy, highlighting the broad applicability of this approach for AMP discovery.
期刊介绍:
The Journal of Chemical Information and Modeling publishes papers reporting new methodology and/or important applications in the fields of chemical informatics and molecular modeling. Specific topics include the representation and computer-based searching of chemical databases, molecular modeling, computer-aided molecular design of new materials, catalysts, or ligands, development of new computational methods or efficient algorithms for chemical software, and biopharmaceutical chemistry including analyses of biological activity and other issues related to drug discovery.
Astute chemists, computer scientists, and information specialists look to this monthly’s insightful research studies, programming innovations, and software reviews to keep current with advances in this integral, multidisciplinary field.
As a subscriber you’ll stay abreast of database search systems, use of graph theory in chemical problems, substructure search systems, pattern recognition and clustering, analysis of chemical and physical data, molecular modeling, graphics and natural language interfaces, bibliometric and citation analysis, and synthesis design and reactions databases.


 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


