D. M. Kovtun, Z. G. Bazhanova, I. F. Shishkov, Y. I. Tarasov
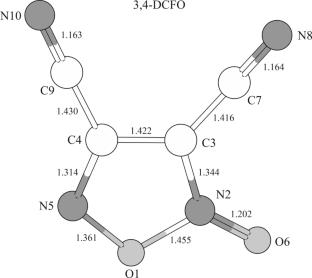
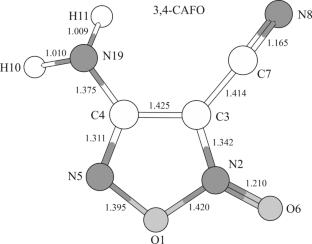
{"title":"Verification of the Experimental Equilibrium Structure of 3,4-Dicyanofuroxan and 3-Cyano-4-Aminofuroxan Molecules by MP2 and CCSD(T) Methods","authors":"D. M. Kovtun, Z. G. Bazhanova, I. F. Shishkov, Y. I. Tarasov","doi":"10.1134/S0022476625080189","DOIUrl":null,"url":null,"abstract":"<p>The equilibrium (<i>r</i><sub><i>e</i></sub>) structure of 3-cyano-4-aminofuroxan (3,4-CAFO) and 3,4-dicyanofuroxan (3,4-DCFO) molecules is determined for the first time by combined QC calculations using post-Hartree–Fock methods (MP2, CCSD(T)) with cc-pV<i>X</i>Z (<i>X</i> = T, Q) basis sets up to the CCSD(T)/cc-pVQZ level of theory. The CCSD(T) single-reference approximation is applied using the Lee–Taylor criterion (<0.02). Theoretical and experimental values of geometric <i>r</i><sub><i>e</i></sub>-parameters of these molecules in the gas phase are compared. QC calculations (CCSD(T)) generally agree with the experimental <i>r</i><sub><i>e</i></sub>-structure of these molecules determined by gas phase electron diffraction (GED). Discrepancies between the values of theoretical and experimental parameters in 3,4-DCFO (4 distances) and 3,4-CAFO (2 distances and 3 angles) molecules are several times higher than the available statistics (CCSD(T)) and the reported experimental errors. The revealed systematic inconsistencies between theoretical and experimental values of some bond lengths and bond angles make these molecules interesting objects for theoretical and experimental structural studies in order to obtain refined geometric <i>r</i><sub><i>e</i></sub>-parameters of furoxan compounds and to accumulate the statistics of <i>r</i><sub><i>e</i></sub>-structures reproduction using high-level quantum chemical methods. When solving the structural inverse problem (IP), it is desirable that balanced molecular models and optimization strategies are selected and applied on a competitive basis.</p>","PeriodicalId":668,"journal":{"name":"Journal of Structural Chemistry","volume":"66 8","pages":"1730 - 1742"},"PeriodicalIF":1.4000,"publicationDate":"2025-08-29","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Structural Chemistry","FirstCategoryId":"92","ListUrlMain":"https://link.springer.com/article/10.1134/S0022476625080189","RegionNum":4,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q4","JCRName":"CHEMISTRY, INORGANIC & NUCLEAR","Score":null,"Total":0}
引用次数: 0
Abstract
The equilibrium (re) structure of 3-cyano-4-aminofuroxan (3,4-CAFO) and 3,4-dicyanofuroxan (3,4-DCFO) molecules is determined for the first time by combined QC calculations using post-Hartree–Fock methods (MP2, CCSD(T)) with cc-pVXZ (X = T, Q) basis sets up to the CCSD(T)/cc-pVQZ level of theory. The CCSD(T) single-reference approximation is applied using the Lee–Taylor criterion (<0.02). Theoretical and experimental values of geometric re-parameters of these molecules in the gas phase are compared. QC calculations (CCSD(T)) generally agree with the experimental re-structure of these molecules determined by gas phase electron diffraction (GED). Discrepancies between the values of theoretical and experimental parameters in 3,4-DCFO (4 distances) and 3,4-CAFO (2 distances and 3 angles) molecules are several times higher than the available statistics (CCSD(T)) and the reported experimental errors. The revealed systematic inconsistencies between theoretical and experimental values of some bond lengths and bond angles make these molecules interesting objects for theoretical and experimental structural studies in order to obtain refined geometric re-parameters of furoxan compounds and to accumulate the statistics of re-structures reproduction using high-level quantum chemical methods. When solving the structural inverse problem (IP), it is desirable that balanced molecular models and optimization strategies are selected and applied on a competitive basis.
期刊介绍:
Journal is an interdisciplinary publication covering all aspects of structural chemistry, including the theory of molecular structure and chemical bond; the use of physical methods to study the electronic and spatial structure of chemical species; structural features of liquids, solutions, surfaces, supramolecular systems, nano- and solid materials; and the crystal structure of solids.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


