Computational insights into hydrogen adsorption energies on medium-entropy oxides
IF 4.2
3区 化学
Q2 CHEMISTRY, PHYSICAL
引用次数: 0
Abstract
High entropy oxides (HEOs) have emerged as promising catalysts for several important chemical transformations including alkane activation. Hydrogen adsorption energy (HAE) has been used as a key descriptor for many reactions including methane C–H activation and hydrogen evolution reactions. Hence, understanding the relationship between HAEs and the surface chemistry of HEO surfaces could lay the foundation for meaningful correlations among methane C–H activation, HAE, and the complex, local environment of HEO surfaces. Here, we used a medium-entropy oxide as a prototypical system – Mg0.25Ni0.25Cu0.25Zn0.25O with a rock-salt structure – to interrogate these relationships. We sampled 2000 different surfaces of its (100) plane and calculated the HAEs at randomly chosen surface O sites using density functional theory (DFT). Our analysis of the 2000 data points reveals that the HAEs at the surface O sites are significantly influenced by the local environment around the adsorption sites, particularly the nature of the metal atom directly below the surface O site where H adsorbs. After comparing several popular graph-neural-network-based machine learning models, we found that the DimeNet++ model performed best achieving satisfactory accuracy in predicting HAEs for both Mg0.25Ni0.25Cu0.25Zn0.25O and slightly varied compositions. Our work underscores the promise of such models and the need for further refinement to address the complexity of HEOs.
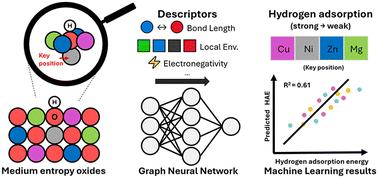
氢在中熵氧化物上吸附能的计算见解
高熵氧化物(HEOs)已成为几种重要化学转化的催化剂,包括烷烃活化。氢吸附能(HAE)是甲烷C-H活化反应和析氢反应的重要描述因子。因此,了解HAEs与HEO表面化学之间的关系可以为甲烷C-H活化、HAE和HEO表面复杂的局部环境之间的有意义的相关性奠定基础。在这里,我们使用中熵氧化物作为原型系统-具有岩盐结构的Mg0.25Ni0.25Cu0.25Zn0.25O -来询问这些关系。我们对其(100)平面的2000个不同表面进行了采样,并使用密度泛函理论(DFT)计算了随机选择的表面0点的HAEs。我们对2000个数据点的分析表明,表面O位点的HAEs受到吸附位点周围的局部环境的显著影响,特别是H吸附的表面O位点正下方金属原子的性质。在比较了几种流行的基于图神经网络的机器学习模型后,我们发现DimeNet++模型在预测Mg0.25Ni0.25Cu0.25Zn0.25O和轻微变化成分的HAEs方面表现最好,取得了令人满意的准确性。我们的工作强调了这些模型的前景,以及进一步改进以解决heo复杂性的必要性。
本文章由计算机程序翻译,如有差异,请以英文原文为准。
求助全文
约1分钟内获得全文
求助全文
来源期刊

Catalysis Science & Technology
CHEMISTRY, PHYSICAL-
CiteScore
8.70
自引率
6.00%
发文量
587
审稿时长
1.5 months
期刊介绍:
A multidisciplinary journal focusing on cutting edge research across all fundamental science and technological aspects of catalysis.
Editor-in-chief: Bert Weckhuysen
Impact factor: 5.0
Time to first decision (peer reviewed only): 31 days
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


