First-principles study of lead-free Na2TmAgCl6 and Na2TmCuCl6 double halide perovskites for photovoltaic and thermoelectric applications
IF 3
4区 生物学
Q2 BIOCHEMICAL RESEARCH METHODS
引用次数: 0
Abstract
This study thoroughly investigates the lead-free halide double perovskites Na2TmAeCl6 (Ae = Ag, Cu) using first-principles density functional theory (DFT) within the CASTEP framework. The formation energies are −3.987 eV for Na2TmAgCl6 and −3.453 eV for Na2TmCuCl6. Additionally, phonon spectra without imaginary frequencies confirm their thermodynamic and dynamic stability. Both compounds adopt a cubic Fm-3m structure with tolerance factors of 0.76 and 0.83, placing them within the stable perovskite range. The elastic constants meet the Born-Huang stability criteria, and Pugh's ratios of 3.48 for Ag and 3.23 for Cu indicate they are ductile. Moreover, Debye temperatures of 124.48 K for Ag and 91.53 K for Cu suggest good lattice thermal stability. Electronic structure analysis shows Na2TmAgCl6 has a direct bandgap of 2.51 eV, while Na2TmCuCl6 features a direct bandgap of 2.32 eV. Their optical properties reveal high absorption coefficients of approximately 16 × 104 cm−1 in the UV region. The calculated SLME efficiencies are 11 % for the Ag compound and 16.5 % for the Cu compound, with ZT values of 0.91 and 1.20, respectively, indicating strong thermoelectric potential. These findings suggest that both Na2TmAgCl6 and Na2TmCuCl6 are promising candidates for advanced optoelectronic, photovoltaic, and thermoelectric applications.
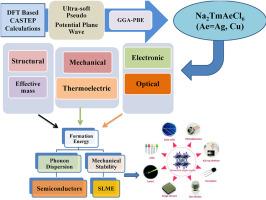
无铅Na2TmAgCl6和Na2TmCuCl6双卤化物钙钛矿的第一性原理研究
本研究在CASTEP框架下使用第一性原理密度泛函理论(DFT)对无铅卤化物双钙钛矿Na2TmAeCl6 (Ae = Ag, Cu)进行了深入研究。Na2TmAgCl6的形成能为- 3.987 eV, Na2TmCuCl6的形成能为- 3.453 eV。此外,没有虚频率的声子谱证实了它们的热力学和动力学稳定性。两种化合物均采用立方的Fm-3m结构,容差系数分别为0.76和0.83,处于稳定的钙钛矿范围内。弹性常数满足Born-Huang稳定性判据,Ag的Pugh比为3.48,Cu的Pugh比为3.23,表明它们具有延展性。此外,银的Debye温度为124.48 K,铜的Debye温度为91.53 K,显示出良好的晶格热稳定性。电子结构分析表明,Na2TmAgCl6的直接带隙为2.51 eV, Na2TmCuCl6的直接带隙为2.32 eV。它们的光学性质显示出在紫外区大约16 × 104 cm−1的高吸收系数。计算得到Ag化合物的SLME效率为11%,Cu化合物的SLME效率为16.5%,ZT分别为0.91和1.20,表明该化合物具有较强的热电势。这些发现表明,Na2TmAgCl6和Na2TmCuCl6都是先进光电、光伏和热电应用的有希望的候选材料。
本文章由计算机程序翻译,如有差异,请以英文原文为准。
求助全文
约1分钟内获得全文
求助全文
来源期刊

Journal of molecular graphics & modelling
生物-计算机:跨学科应用
CiteScore
5.50
自引率
6.90%
发文量
216
审稿时长
35 days
期刊介绍:
The Journal of Molecular Graphics and Modelling is devoted to the publication of papers on the uses of computers in theoretical investigations of molecular structure, function, interaction, and design. The scope of the journal includes all aspects of molecular modeling and computational chemistry, including, for instance, the study of molecular shape and properties, molecular simulations, protein and polymer engineering, drug design, materials design, structure-activity and structure-property relationships, database mining, and compound library design.
As a primary research journal, JMGM seeks to bring new knowledge to the attention of our readers. As such, submissions to the journal need to not only report results, but must draw conclusions and explore implications of the work presented. Authors are strongly encouraged to bear this in mind when preparing manuscripts. Routine applications of standard modelling approaches, providing only very limited new scientific insight, will not meet our criteria for publication. Reproducibility of reported calculations is an important issue. Wherever possible, we urge authors to enhance their papers with Supplementary Data, for example, in QSAR studies machine-readable versions of molecular datasets or in the development of new force-field parameters versions of the topology and force field parameter files. Routine applications of existing methods that do not lead to genuinely new insight will not be considered.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


