In silico design of endothelin receptor antagonists using Monte Carlo-based QSAR modeling, molecular docking, and ADME profiling
IF 3
4区 生物学
Q2 BIOCHEMICAL RESEARCH METHODS
引用次数: 0
Abstract
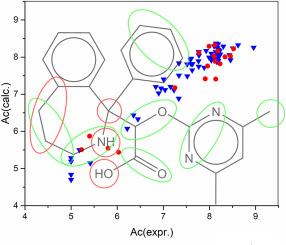
Endothelin-1 (ET-1), a potent vasoconstrictor peptide, plays a critical role in cardiovascular pathologies and remains a key target for therapeutic intervention. Despite its clinical significance, the development of selective and potent ET-1 antagonists continues to present major challenges. Computational methods, particularly Quantitative Structure–Activity Relationship (QSAR) modeling, offer a rational and efficient framework for designing such compounds. In this study, conformation-independent QSAR models were developed using molecular descriptors derived from SMILES notation and local molecular graph invariants. The Monte Carlo method was employed for descriptor selection and weight optimization, resulting in statistically robust models. Critical molecular fragments associated with antagonist activity were identified and applied in the computer-aided design (CAD) of new ET-1 inhibitors. The optimal QSAR model exhibited strong predictive performance, with high correlation coefficients for both the training set (r2 = 0.9362, q2 = 0.9314) and the test set (r2 = 0.9006, q2 = 0.8655). To further validate the structural plausibility of the designed molecules, molecular docking simulations were conducted against the ETA receptor. The docking results were in agreement with QSAR-predicted activity, revealing favorable binding poses, strong interaction energies, and consistent structure–activity trends across all six designed compounds. This methodological convergence strengthens the credibility of the in silico predictions. Additionally, computational analysis of physicochemical and pharmacokinetic parameters indicated favorable ADME profiles, high drug-likeness, and efficient gastrointestinal absorption, suggesting suitability for medicinal chemistry development. This study introduces a reliable and mechanistically interpretable computational pipeline for the discovery of novel ET-1 antagonists. The proposed compounds demonstrate promising pharmacological characteristics and represent viable candidates for future experimental validation. These findings underscore the value of integrated in silico strategies in accelerating cardiovascular drug discovery.
基于蒙特卡罗的QSAR建模、分子对接和ADME分析的内皮素受体拮抗剂的计算机设计
内皮素-1 (ET-1)是一种有效的血管收缩肽,在心血管疾病中起着关键作用,是治疗干预的关键靶点。尽管具有临床意义,但选择性和有效的ET-1拮抗剂的开发仍然面临重大挑战。计算方法,特别是定量构效关系(QSAR)模型,为此类化合物的设计提供了合理有效的框架。在本研究中,利用源自SMILES符号和局部分子图不变量的分子描述符,建立了与构象无关的QSAR模型。采用蒙特卡罗方法进行描述符的选择和权值的优化,得到了具有统计鲁棒性的模型。确定了与拮抗剂活性相关的关键分子片段,并将其应用于新的ET-1抑制剂的计算机辅助设计(CAD)中。最优QSAR模型对训练集(r2 = 0.9362, q2 = 0.9314)和检验集(r2 = 0.9006, q2 = 0.8655)均具有较高的相关系数,具有较强的预测能力。为了进一步验证所设计分子的结构合理性,针对ETA受体进行了分子对接模拟。对接结果与qsar预测的活性一致,揭示了所有六个设计化合物的有利结合姿态,强相互作用能和一致的结构-活性趋势。这种方法的趋同增强了计算机预测的可信度。此外,理化和药代动力学参数的计算分析表明,ADME谱良好,药物相似度高,胃肠道吸收效率高,适合药物化学开发。这项研究为发现新的ET-1拮抗剂引入了一个可靠的、机械上可解释的计算管道。所提出的化合物显示出有希望的药理特性,并代表了未来实验验证的可行候选物。这些发现强调了集成芯片策略在加速心血管药物发现方面的价值。
本文章由计算机程序翻译,如有差异,请以英文原文为准。
求助全文
约1分钟内获得全文
求助全文
来源期刊

Journal of molecular graphics & modelling
生物-计算机:跨学科应用
CiteScore
5.50
自引率
6.90%
发文量
216
审稿时长
35 days
期刊介绍:
The Journal of Molecular Graphics and Modelling is devoted to the publication of papers on the uses of computers in theoretical investigations of molecular structure, function, interaction, and design. The scope of the journal includes all aspects of molecular modeling and computational chemistry, including, for instance, the study of molecular shape and properties, molecular simulations, protein and polymer engineering, drug design, materials design, structure-activity and structure-property relationships, database mining, and compound library design.
As a primary research journal, JMGM seeks to bring new knowledge to the attention of our readers. As such, submissions to the journal need to not only report results, but must draw conclusions and explore implications of the work presented. Authors are strongly encouraged to bear this in mind when preparing manuscripts. Routine applications of standard modelling approaches, providing only very limited new scientific insight, will not meet our criteria for publication. Reproducibility of reported calculations is an important issue. Wherever possible, we urge authors to enhance their papers with Supplementary Data, for example, in QSAR studies machine-readable versions of molecular datasets or in the development of new force-field parameters versions of the topology and force field parameter files. Routine applications of existing methods that do not lead to genuinely new insight will not be considered.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


