Antiplasmodial activity of pentyloxyamide-based histone deacetylase inhibitors against Plasmodium falciparum parasites
IF 3.4
2区 医学
Q1 PARASITOLOGY
International Journal for Parasitology: Drugs and Drug Resistance
Pub Date : 2025-08-16
DOI:10.1016/j.ijpddr.2025.100608
引用次数: 0
Abstract
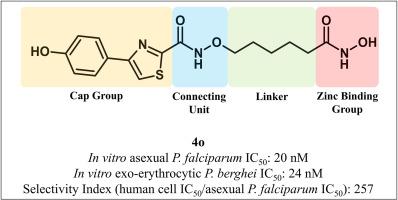
Malaria is caused by Plasmodium parasites and remains a significant health concern for almost half the world's population. There are estimated to be > 240 million malaria cases and approximately 600,000 malaria-related deaths annually, mainly due to infection with P. falciparum parasites. Parasite drug resistance is impacting malaria prevention and control efforts, and as part of the malaria eradication agenda, new drugs with novel mechanisms of action are needed. Histone/lysine deacetylase (HDAC) enzymes play essential roles in Plasmodium biology and are potential targets for the development of new antiplasmodial agents. In this study, a panel of 24 HDAC inhibitors with hydroxamic acid zinc binding group, a pentyloxyamide connecting unit linker region and substituted 4-phenyl and 4(pyridinyl)thiazole cap groups were investigated for in vitro activity against asexual intraerythrocytic stage P. falciparum parasites, the life cycle stage responsible for the clinical symptoms of malaria. The most potent compound (4o) had a P. falciparum IC50 of 20 nM and >250-fold greater selectivity for P. falciparum versus human cells. Compound 4o was also active against exoerythrocytic stage parasites (IC50 24 nM), which are a target for malaria prevention. In contrast, 4o lacked potent activity against late-stage gametocytes (IC50 > 2 μM), which are a target for malaria transmission-blocking drugs. Compound 4o and analogues caused in situ hyperacetylation of P. falciparum histone H4, indicating deacetylase inhibition. Furthermore, 4o was found to stabilise PfHDAC1 in P. falciparum protein lysates using solvent-induced protein stability Western blot assays with anti-PfHDAC1 antibody. Together, these data provide new structure-activity relationship and mechanistic insights on pentyloxyamide-based HDAC inhibitors as potential therapeutic starting points for malaria.
戊酰氧胺基组蛋白去乙酰酶抑制剂对恶性疟原虫的抗疟原虫活性研究
疟疾是由疟原虫引起的,对世界上几乎一半的人口来说仍然是一个重大的健康问题。估计每年有2.4亿疟疾病例和大约60万与疟疾有关的死亡,主要是由于感染恶性疟原虫。寄生虫耐药性正在影响疟疾的预防和控制工作,作为消灭疟疾议程的一部分,需要具有新的作用机制的新药。组蛋白/赖氨酸脱乙酰酶(HDAC)酶在疟原虫生物学中起着重要作用,是开发新的抗疟原虫药物的潜在靶点。在这项研究中,研究了24种HDAC抑制剂的体外活性,它们具有羟肟酸锌结合基团、戊氧基酰胺连接单元连接区和取代4-苯基和4(吡啶基)噻唑帽基团,以对抗红细胞内无性恶性疟原虫寄生虫,这是导致疟疾临床症状的生命周期阶段。最有效的化合物(40)对恶性疟原虫的IC50为20 nM,对恶性疟原虫的选择性比人类细胞高250倍。化合物40对体外红细胞期寄生虫(IC50为24 nM)也有活性,这是疟疾预防的靶点。相比之下,40缺乏对晚期配子体(IC50 > 2 μM)的有效活性,后者是疟疾传播阻断药物的靶点。化合物40和类似物引起恶性疟原虫组蛋白H4原位高乙酰化,表明去乙酰化酶抑制。此外,使用抗PfHDAC1抗体的溶剂诱导蛋白稳定性Western blot检测,发现40在恶性疟原虫蛋白溶出物中稳定PfHDAC1。总之,这些数据提供了基于戊氧胺的HDAC抑制剂作为疟疾潜在治疗起点的新的结构-活性关系和机制见解。
本文章由计算机程序翻译,如有差异,请以英文原文为准。
求助全文
约1分钟内获得全文
求助全文
来源期刊

International Journal for Parasitology: Drugs and Drug Resistance
PARASITOLOGY-PHARMACOLOGY & PHARMACY
CiteScore
7.90
自引率
7.50%
发文量
31
审稿时长
48 days
期刊介绍:
The International Journal for Parasitology – Drugs and Drug Resistance is one of a series of specialist, open access journals launched by the International Journal for Parasitology. It publishes the results of original research in the area of anti-parasite drug identification, development and evaluation, and parasite drug resistance. The journal also covers research into natural products as anti-parasitic agents, and bioactive parasite products. Studies can be aimed at unicellular or multicellular parasites of human or veterinary importance.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


