Identification of novel non-covalent Mpro inhibitors against SARS-CoV-2 by the computational and experimental approaches
IF 3
4区 生物学
Q2 BIOCHEMICAL RESEARCH METHODS
引用次数: 0
Abstract
The COVID-19 pandemic, caused by SARS-CoV-2, has posed significant global health challenges and there is an urgent need for effective therapeutic agents. The main protease (Mpro) plays a crucial role in viral replication, making it an attractive target for the development of antiviral drugs. In this study, by screening over 8.06 million compounds obtained from Enamine, Vitas-M, ChemDiv, and TargetMol (USA) databases, 52 top-ranking compounds were obtained as promising candidates through molecular docking, followed by Uni-QSAR modeling and ADMET predictions to evaluate their binding affinities and pharmacokinetic properties. Biological activity assays confirmed the efficacy of four standout candidates L17, L26, L37, and L50 with IC50 values of 5.61 ± 0.58 μM, 6.00 ± 0.63 μM, 4.21 ± 0.89 μM, and 2.84 ± 1.20 μM, respectively, comparable to that of reference ML188 (2.41 ± 0.70 μM). Further insights were gained through density functional theory (DFT) analyses, which provided valuable information regarding the electronic and structural properties of the candidate compounds. Additionally, extensive molecular dynamics (MD) simulations were conducted, revealing critical information about the stability and binding interactions of the compounds within the Mpro active site over a 500 ns simulation period. Besides, the results of binding free energy calculations demonstrated that compounds had higher binding affinity than ML188, and dcTMD simulations further revealed that L26, L37, and L50 followed more favorable and cooperative unbinding pathways with higher energy barriers and lower dissipation compared to ML188. Overall, the results highlight the therapeutic potential of these compounds as effective Mpro inhibitors, laying a solid foundation for further development into novel antiviral agents against SARS-CoV-2.
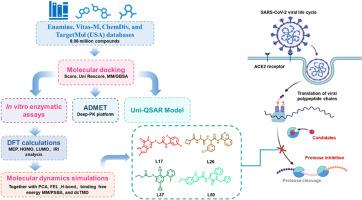
通过计算和实验方法鉴定抗SARS-CoV-2的新型非共价Mpro抑制剂
由SARS-CoV-2引起的COVID-19大流行给全球卫生带来了重大挑战,迫切需要有效的治疗药物。主蛋白酶(Mpro)在病毒复制中起着至关重要的作用,使其成为抗病毒药物开发的一个有吸引力的靶点。本研究通过对来自Enamine、Vitas-M、ChemDiv和TargetMol (USA)数据库的806万种化合物进行筛选,通过分子对接,通过Uni-QSAR建模和ADMET预测,获得了52种排名最高的候选化合物,并评估了它们的结合亲和力和药代动力学性质。生物活性实验证实,L17、L26、L37和L50的IC50值分别为5.61±0.58 μM、6.00±0.63 μM、4.21±0.89 μM和2.84±1.20 μM,与参比物ML188的IC50值(2.41±0.70 μM)相当。通过密度泛函理论(DFT)分析获得了进一步的见解,这为候选化合物的电子和结构特性提供了有价值的信息。此外,进行了广泛的分子动力学(MD)模拟,揭示了500 ns模拟周期内Mpro活性位点内化合物的稳定性和结合相互作用的关键信息。此外,结合自由能计算结果表明,化合物的结合亲和力高于ML188, dcTMD模拟结果进一步表明,与ML188相比,L26、L37和L50具有更高的能量势垒和更低的耗散,遵循更有利的合作解结合途径。总的来说,这些结果突出了这些化合物作为有效的Mpro抑制剂的治疗潜力,为进一步开发针对SARS-CoV-2的新型抗病毒药物奠定了坚实基础。
本文章由计算机程序翻译,如有差异,请以英文原文为准。
求助全文
约1分钟内获得全文
求助全文
来源期刊

Journal of molecular graphics & modelling
生物-计算机:跨学科应用
CiteScore
5.50
自引率
6.90%
发文量
216
审稿时长
35 days
期刊介绍:
The Journal of Molecular Graphics and Modelling is devoted to the publication of papers on the uses of computers in theoretical investigations of molecular structure, function, interaction, and design. The scope of the journal includes all aspects of molecular modeling and computational chemistry, including, for instance, the study of molecular shape and properties, molecular simulations, protein and polymer engineering, drug design, materials design, structure-activity and structure-property relationships, database mining, and compound library design.
As a primary research journal, JMGM seeks to bring new knowledge to the attention of our readers. As such, submissions to the journal need to not only report results, but must draw conclusions and explore implications of the work presented. Authors are strongly encouraged to bear this in mind when preparing manuscripts. Routine applications of standard modelling approaches, providing only very limited new scientific insight, will not meet our criteria for publication. Reproducibility of reported calculations is an important issue. Wherever possible, we urge authors to enhance their papers with Supplementary Data, for example, in QSAR studies machine-readable versions of molecular datasets or in the development of new force-field parameters versions of the topology and force field parameter files. Routine applications of existing methods that do not lead to genuinely new insight will not be considered.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


