Design, development, and testing of a new multi-locus sequence typing scheme for the zoonotic pathogen Cryptosporidium parvum
IF 1.7
Q3 PARASITOLOGY
Current research in parasitology & vector-borne diseases
Pub Date : 2025-01-01
DOI:10.1016/j.crpvbd.2025.100308
引用次数: 0
Abstract
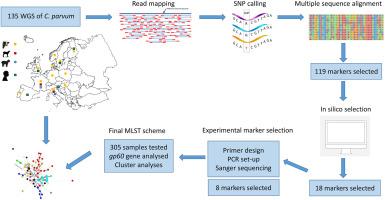
The zoonotic parasite Cryptosporidium parvum is an important global cause of diarrheal disease in humans and young ruminants. Molecular typing is essential to track transmission routes and identify clusters of cases. Here, we developed a novel multi-locus sequence typing (MLST) scheme based on single nucleotide polymorphisms (SNPs) in unlinked markers. Coding regions with high variability were identified by comparing whole genome sequences (WGS) from 43 human- and 92 ruminant-derived C. parvum samples collected across Europe. We first selected 18 markers and showed that they provide high discrimination among the samples with WGS data, with 88% of the MLSTs being singletons. Next, we defined a MLST based on eight genetically unlinked markers and generated sequence data from 305 C. parvum samples, collected from four different host species and 13 European countries. We consolidated a set of 365 fully genotyped samples, characterized by the presence of 154 different MLSTs, 105 of which were singletons. Network analyses showed no complete clustering of samples by host species or country of origin at the European scale. We further showed that samples with gp60 subtypes that are common in Europe are divided into many MLSTs by the new scheme, highlighting its increased discriminatory ability. However, the applicability of the scheme in public health settings is limited by its cost, turnaround time, and scalability. To achieve discrimination of C. parvum samples based on SNPs, a large number of loci needs to be analysed, and this is feasible using amplicon sequencing technologies.
一种新的人畜共患病原体小隐孢子虫多位点序列分型方案的设计、开发和测试
人畜共患寄生虫小隐孢子虫是人类和幼龄反刍动物腹泻病的重要全球病因。分子分型对于追踪传播途径和确定聚集性病例至关重要。在这里,我们开发了一种新的基于非连锁标记的单核苷酸多态性(snp)的多位点序列分型(MLST)方案。通过比较来自欧洲各地收集的43个人类和92个反刍动物来源的小弧菌样本的全基因组序列(WGS),确定了具有高变异性的编码区。我们首先选择了18个标记,并表明它们在WGS数据的样本中具有很高的区分性,其中88%的mlst是单基因。接下来,我们基于8个基因不连锁的标记定义了一个MLST,并生成了来自4个不同宿主物种和13个欧洲国家的305个小孢子虫样本的序列数据。我们整合了365份完全基因分型的样本,其特征是存在154个不同的mlst,其中105个为单基因。网络分析显示,在欧洲范围内,按宿主物种或原产国划分的样本没有完全聚类。我们进一步发现,在欧洲常见的gp60亚型样本被新方案划分为许多mlst,突出了其增加的区分能力。然而,该方案在公共卫生环境中的适用性受到其成本、周转时间和可扩展性的限制。为了实现基于snp的小弧菌样本的区分,需要分析大量的位点,使用扩增子测序技术是可行的。
本文章由计算机程序翻译,如有差异,请以英文原文为准。
求助全文
约1分钟内获得全文
求助全文

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


