{"title":"FAK inhibition disrupts tumor growth, apoptosis, and transcriptional regulation in GI-NETs.","authors":"Lara Toffoli, Angeliki Ditsiou, Luca Triboli, Victorine Hamm, Eva Moschioni, Francesca D'Este, Teresa Gagliano","doi":"10.1530/EO-25-0052","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>Gastrointestinal neuroendocrine tumors (GI-NETs) are rare neoplasms with limited therapeutic options and increasing clinical incidence. Focal adhesion kinase (FAK) has been implicated in oncogenic processes across various tumor types; however, its specific role in GI-NET biology remains inadequately characterized. This study investigates the impact of FAK inhibition on GI-NET cell survival, invasive potential, and gene regulation, with the aim of evaluating FAK as a therapeutic target.</p><p><strong>Methods: </strong>Human GI-NET cell lines (GOT1 and COLO320DM) were treated with Y15, a kinase inhibitor, and PROTAC-FAK (BI-0319), a degrader that abrogates both enzymatic and scaffold functions. siRNA-mediated knockdown of FAK was employed for functional validation. Assays assessing viability and apoptosis were performed in both 2D and 3D culture conditions, while invasion and colony formation were assessed in 2D culture. Western blotting, immunofluorescence, and qRT-PCR were used to evaluate molecular effects. Public transcriptomic datasets were analyzed to assess PTK2 expression across NET subtypes.</p><p><strong>Results: </strong>FAK inhibition reduced cell viability, colony formation, and invasive capacity. PROTAC-FAK, but not Y15, decreased H3K9 acetylation, indicating scaffold-dependent epigenetic modulation. On the other hand, both PROTAC-FAK and Y15 decreased H3K4 methylation levels, further supporting the role of FAK in chromatin regulation. Both compounds suppressed ERK1/2 phosphorylation and modulated RB1 expression, which was further validated by FAK knockdown. In silico analysis revealed elevated PTK2 expression in rectal and small intestinal NETs relative to pancreatic NETs.</p><p><strong>Conclusion: </strong>These findings identify FAK as a regulator of oncogenic and epigenetic pathways in GI-NETs and support its therapeutic targeting, particularly through degradation strategies that inhibit its non-catalytic functions.</p><p><strong>Highlights: </strong>FAK inhibition impairs GI-NET viability, invasion, and colony formation in both 2D and 3D models using kinase (Y15) and PROTAC-based degraders (BI-0319).PROTAC-FAK uniquely reduces H3K9 acetylation, revealing a kinase-independent scaffold role for FAK in epigenetic regulation.esiRNA knockdown of FAK recapitulates pharmacological effects, confirming FAK as a driver of oncogenic features in GI-NET cells.In silico analysis identifies elevated PTK2 expression in rectal and small intestine NETs, with a strong positive correlation to RB1, supporting translational relevance.</p>","PeriodicalId":72907,"journal":{"name":"Endocrine oncology (Bristol, England)","volume":"5 1","pages":"e250052"},"PeriodicalIF":0.0000,"publicationDate":"2025-08-14","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12358824/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Endocrine oncology (Bristol, England)","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1530/EO-25-0052","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2025/1/1 0:00:00","PubModel":"eCollection","JCR":"","JCRName":"","Score":null,"Total":0}
引用次数: 0
Abstract
Background: Gastrointestinal neuroendocrine tumors (GI-NETs) are rare neoplasms with limited therapeutic options and increasing clinical incidence. Focal adhesion kinase (FAK) has been implicated in oncogenic processes across various tumor types; however, its specific role in GI-NET biology remains inadequately characterized. This study investigates the impact of FAK inhibition on GI-NET cell survival, invasive potential, and gene regulation, with the aim of evaluating FAK as a therapeutic target.
Methods: Human GI-NET cell lines (GOT1 and COLO320DM) were treated with Y15, a kinase inhibitor, and PROTAC-FAK (BI-0319), a degrader that abrogates both enzymatic and scaffold functions. siRNA-mediated knockdown of FAK was employed for functional validation. Assays assessing viability and apoptosis were performed in both 2D and 3D culture conditions, while invasion and colony formation were assessed in 2D culture. Western blotting, immunofluorescence, and qRT-PCR were used to evaluate molecular effects. Public transcriptomic datasets were analyzed to assess PTK2 expression across NET subtypes.
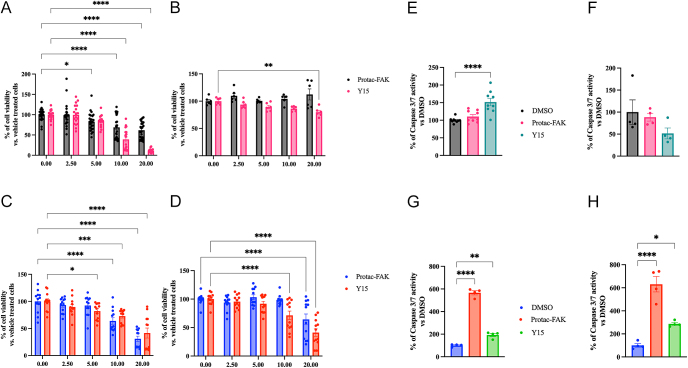
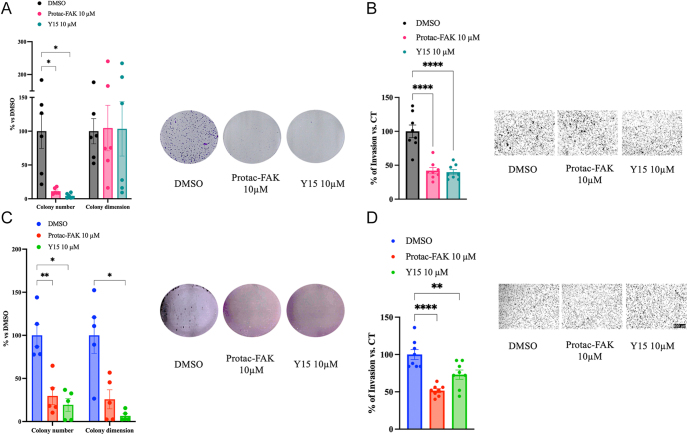
Results: FAK inhibition reduced cell viability, colony formation, and invasive capacity. PROTAC-FAK, but not Y15, decreased H3K9 acetylation, indicating scaffold-dependent epigenetic modulation. On the other hand, both PROTAC-FAK and Y15 decreased H3K4 methylation levels, further supporting the role of FAK in chromatin regulation. Both compounds suppressed ERK1/2 phosphorylation and modulated RB1 expression, which was further validated by FAK knockdown. In silico analysis revealed elevated PTK2 expression in rectal and small intestinal NETs relative to pancreatic NETs.
Conclusion: These findings identify FAK as a regulator of oncogenic and epigenetic pathways in GI-NETs and support its therapeutic targeting, particularly through degradation strategies that inhibit its non-catalytic functions.
Highlights: FAK inhibition impairs GI-NET viability, invasion, and colony formation in both 2D and 3D models using kinase (Y15) and PROTAC-based degraders (BI-0319).PROTAC-FAK uniquely reduces H3K9 acetylation, revealing a kinase-independent scaffold role for FAK in epigenetic regulation.esiRNA knockdown of FAK recapitulates pharmacological effects, confirming FAK as a driver of oncogenic features in GI-NET cells.In silico analysis identifies elevated PTK2 expression in rectal and small intestine NETs, with a strong positive correlation to RB1, supporting translational relevance.


 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


