Mining the plasma proteome: Evaluation of enrichment methods for depth and reproducibility
IF 2.8
2区 生物学
Q2 BIOCHEMICAL RESEARCH METHODS
引用次数: 0
Abstract
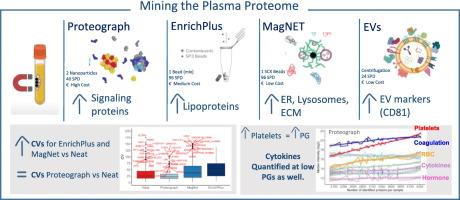
Plasma is a complex biological fluid containing extracellular vesicles (EVs), residual platelets, and soluble proteins. While conventional plasma proteomics typically identifies hundreds of proteins, recent enrichment strategies have expanded coverage to thousands. It is still unclear whether these methods enrich preferentially different classes of protein and whether they allow for reliable quantification. Here, we compared three common advanced proteomic workflows—Proteograph (Seer), Mag-Net (ReSynBio), and ENRICHplus (PreOmics) as well as EV enrichment obtained by centrifugation. We explore the content in soluble proteins, EV cargo, and platelet-derived proteins after the enrichments. Quantification was evaluated comparing each method to neat plasma using protein coefficient of variation and point-biserial correlation.
We quantified an average of ∼4500 proteins with EV centrifugation, ∼4000 with Seer, ∼2800 with ENRICHplus, ∼2300 with Mag-Net, and ∼ 900 with neat plasma. Each method enriched distinct sets of protein signatures: EV preparations were enriched with EV markers such as CD81; ENRICHplus predominantly captured lipoproteins; and Proteograph was enriched for cytokines and hormones. Platelet protein intensity was directly correlated with total protein identifications but did not compromise quantification of low-abundance proteins. Across 50 healthy individuals, Proteograph consistently demonstrated reproducible enrichment and depletion patterns, with some reported exceptions.
Our results highlight the strengths and biases of different plasma enrichment strategies.
Significance
This study benchmarks corona formation strategies for enriching low-abundance plasma proteins, including those from platelets and EVs. While enabling deeper proteome coverage compared to depletion methods, these approaches also reshape the intensity landscape and reveal method-specific patterns in protein class enrichment and in quantification repeatability.
血浆蛋白质组的挖掘:深度和可重复性富集方法的评价。
血浆是一种复杂的生物液体,含有细胞外囊泡(EVs)、残留的血小板和可溶性蛋白质。虽然传统的血浆蛋白质组学通常识别数百种蛋白质,但最近的富集策略已将覆盖范围扩大到数千种。目前尚不清楚这些方法是否优先富集不同种类的蛋白质,以及它们是否允许可靠的定量。在这里,我们比较了三种常见的先进蛋白质组学工作流程- proteograph (Seer), magg - net (ReSynBio)和enrichment plus (PreOmics)以及通过离心获得的EV富集。我们研究了富集后可溶性蛋白、EV货物和血小板衍生蛋白的含量。用蛋白质变异系数和点双列相关性对每种方法与纯血浆的定量进行比较。我们用EV离心平均定量了~4500个蛋白,用Seer定量了~4000个,用enrichment定量了~2800个,用Mag-Net定量了~2300个,用纯等离子定量了 ~ 900个。每种方法都富集了不同的蛋白质特征集:EV制剂富集了EV标记物,如CD81;富集主要捕获脂蛋白;蛋白图富含细胞因子和激素。血小板蛋白强度与总蛋白鉴定直接相关,但不影响低丰度蛋白的定量。在50个健康个体中,Proteograph一致显示出可重复的富集和耗尽模式,有一些报道的例外。我们的结果突出了不同等离子体富集策略的优势和偏差。意义:本研究为富集低丰度血浆蛋白(包括来自血小板和ev的蛋白)的冠状形成策略提供了基准。与耗尽方法相比,这些方法能够实现更深的蛋白质组覆盖,同时也重塑了强度景观,并揭示了蛋白质类富集和定量可重复性的方法特异性模式。
本文章由计算机程序翻译,如有差异,请以英文原文为准。
求助全文
约1分钟内获得全文
求助全文
来源期刊

Journal of proteomics
生物-生化研究方法
CiteScore
7.10
自引率
3.00%
发文量
227
审稿时长
73 days
期刊介绍:
Journal of Proteomics is aimed at protein scientists and analytical chemists in the field of proteomics, biomarker discovery, protein analytics, plant proteomics, microbial and animal proteomics, human studies, tissue imaging by mass spectrometry, non-conventional and non-model organism proteomics, and protein bioinformatics. The journal welcomes papers in new and upcoming areas such as metabolomics, genomics, systems biology, toxicogenomics, pharmacoproteomics.
Journal of Proteomics unifies both fundamental scientists and clinicians, and includes translational research. Suggestions for reviews, webinars and thematic issues are welcome.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


