Quantum chemical calculations based on 4,5-di(1,2,4-oxadiazol-3-yl)-2H-1,2,3-triazole and 3,3′ -(2H-1,2,3-triazole-4,5-diyl)-bis(1,2,4-oxadiazol-5(4H)-one) derivatives: a DFT study
Yang Zhu, Peng Zhang, YuQin Chu, Wen Jiang, Peng Ma, CongMing Ma
{"title":"Quantum chemical calculations based on 4,5-di(1,2,4-oxadiazol-3-yl)-2H-1,2,3-triazole and 3,3′ -(2H-1,2,3-triazole-4,5-diyl)-bis(1,2,4-oxadiazol-5(4H)-one) derivatives: a DFT study","authors":"Yang Zhu, Peng Zhang, YuQin Chu, Wen Jiang, Peng Ma, CongMing Ma","doi":"10.1007/s00894-025-06473-x","DOIUrl":null,"url":null,"abstract":"<div><h3>Context</h3><p>1,2,3-Triazole has excellent thermal stability, and both 1,2,3-triazole and 1,2,4-oxazolium are good in structural modifiability. This study employed density functional theory (DFT) to design 24 target compounds (designated <b>PA-1</b> to <b>PA-24</b>) based on the characteristics of these two heterocyclic systems. Using 4,5-diallyl-2H-1,2,3-triazole-3,3′-(2H-1,2,3-triazole-4,5-diyl)-bis(1,2,4-oxadiazol-5(4H)-one) as the parent compound (labeled <b>A1</b>), we introduced different energetic substituents at the C3 position of the oxadiazole ring and the N2 position of the triazole ring. Alternatively, using 3,3′-(2H-1,2,3-triazole-4,5-diyl)-bis(1,2,4-oxadiazol-5(4H)-one) as the parent compound (labeled <b>A2</b>), we introduced different energetic substituents at the N2 position of the triazole ring. The selection process considered both macroscopic properties (heat of formation and detonation performance) and microscopic factors including frontier molecular orbitals, energy gaps, weak interactions, surface electrostatic potential, and bond parameters. This approach aimed to identify energetic materials with both detonation performance and safety characteristics while investigating how introducing different substituents at these two positions affects detonation performance and safety. The research results indicate the following: First, introducing an azide group at the C3 position of the oxadiazole in <b>A1</b> more effectively enhances the compound’s heat of formation. <b>PA-13</b> (△<i>H</i><sub>f,sold</sub> = 972.97 kJ/mol), <b>PA-14</b> (△<i>H</i><sub>f,sold</sub> = 1023.62 kJ/mol), and <b>PA-15</b> (△<i>H</i><sub>f,sold</sub> = 1069.24 kJ/mol), which all contain azide groups, rank among the top three in heat of formation among the 24 compounds. Second, <b>PA-6</b> (<i>D</i> = 9.31 km/s, <i>P</i> = 39.93 GPa, <i>Q</i> = 7.39 kJ/g) exhibits the best detonation performance: its detonation pressure exceeds those of RDX (<i>G</i> = 36GPa), its detonation velocity and detonation heat surpass that of HMX (<i>D</i> = 9 km/s, <i>Q</i> = 6.4 kJ/g), and it achieves the ideal state of zero oxygen balance, resulting in the highest energy utilization efficiency. Third, <b>PA-19</b> showed the least impact sensitivity and showed the potential of an insensitive explosive. Fourth, hydrogen bonds are common in the molecular structure of <b>PA-8</b> ~ <b>PA-12</b> derivatives of <b>A2</b>, which may contribute to the stability of the molecules. Fifth, trinitromethyl and dinitromethyl groups are more prone to fragmentation. The substituents introduced at the C3 position of the oxadiazole have almost no effect on the ring length of the oxadiazole, but introducing trinitromethyl at the N2 position of the triazole causes the triazole ring structure to expand, resulting in an increased ring length.</p><h3>Method</h3><p>All calculations in this paper are based on density functional theory and were performed using the Gaussian16 software. Initially, the structures of <b>PA-1</b> to <b>PA-24</b> were optimized at the B3LYP-D3/6-311G** level. Subsequently, single-point energy calculations were conducted at the M06-2X-D3/def2-TZVPP level to determine the formation enthalpy, detonation velocity, and detonation pressure of the compounds. The Multiwfn and VMD programs were used to plot the energy gap diagrams, isosurface maps, scatter plots, and surface electrostatic potential maps for <b>PA-1</b> to <b>PA-24</b>.</p></div>","PeriodicalId":651,"journal":{"name":"Journal of Molecular Modeling","volume":"31 9","pages":""},"PeriodicalIF":2.5000,"publicationDate":"2025-08-21","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Molecular Modeling","FirstCategoryId":"92","ListUrlMain":"https://link.springer.com/article/10.1007/s00894-025-06473-x","RegionNum":4,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q4","JCRName":"BIOCHEMISTRY & MOLECULAR BIOLOGY","Score":null,"Total":0}
引用次数: 0
Abstract
Context
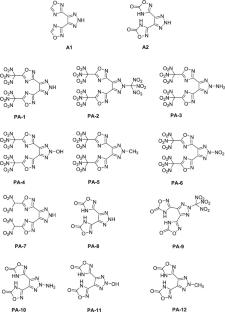
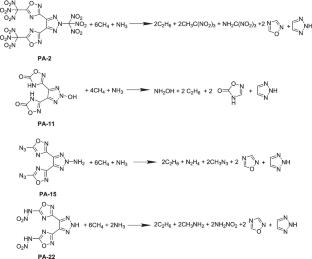
1,2,3-Triazole has excellent thermal stability, and both 1,2,3-triazole and 1,2,4-oxazolium are good in structural modifiability. This study employed density functional theory (DFT) to design 24 target compounds (designated PA-1 to PA-24) based on the characteristics of these two heterocyclic systems. Using 4,5-diallyl-2H-1,2,3-triazole-3,3′-(2H-1,2,3-triazole-4,5-diyl)-bis(1,2,4-oxadiazol-5(4H)-one) as the parent compound (labeled A1), we introduced different energetic substituents at the C3 position of the oxadiazole ring and the N2 position of the triazole ring. Alternatively, using 3,3′-(2H-1,2,3-triazole-4,5-diyl)-bis(1,2,4-oxadiazol-5(4H)-one) as the parent compound (labeled A2), we introduced different energetic substituents at the N2 position of the triazole ring. The selection process considered both macroscopic properties (heat of formation and detonation performance) and microscopic factors including frontier molecular orbitals, energy gaps, weak interactions, surface electrostatic potential, and bond parameters. This approach aimed to identify energetic materials with both detonation performance and safety characteristics while investigating how introducing different substituents at these two positions affects detonation performance and safety. The research results indicate the following: First, introducing an azide group at the C3 position of the oxadiazole in A1 more effectively enhances the compound’s heat of formation. PA-13 (△Hf,sold = 972.97 kJ/mol), PA-14 (△Hf,sold = 1023.62 kJ/mol), and PA-15 (△Hf,sold = 1069.24 kJ/mol), which all contain azide groups, rank among the top three in heat of formation among the 24 compounds. Second, PA-6 (D = 9.31 km/s, P = 39.93 GPa, Q = 7.39 kJ/g) exhibits the best detonation performance: its detonation pressure exceeds those of RDX (G = 36GPa), its detonation velocity and detonation heat surpass that of HMX (D = 9 km/s, Q = 6.4 kJ/g), and it achieves the ideal state of zero oxygen balance, resulting in the highest energy utilization efficiency. Third, PA-19 showed the least impact sensitivity and showed the potential of an insensitive explosive. Fourth, hydrogen bonds are common in the molecular structure of PA-8 ~ PA-12 derivatives of A2, which may contribute to the stability of the molecules. Fifth, trinitromethyl and dinitromethyl groups are more prone to fragmentation. The substituents introduced at the C3 position of the oxadiazole have almost no effect on the ring length of the oxadiazole, but introducing trinitromethyl at the N2 position of the triazole causes the triazole ring structure to expand, resulting in an increased ring length.
Method
All calculations in this paper are based on density functional theory and were performed using the Gaussian16 software. Initially, the structures of PA-1 to PA-24 were optimized at the B3LYP-D3/6-311G** level. Subsequently, single-point energy calculations were conducted at the M06-2X-D3/def2-TZVPP level to determine the formation enthalpy, detonation velocity, and detonation pressure of the compounds. The Multiwfn and VMD programs were used to plot the energy gap diagrams, isosurface maps, scatter plots, and surface electrostatic potential maps for PA-1 to PA-24.
期刊介绍:
The Journal of Molecular Modeling focuses on "hardcore" modeling, publishing high-quality research and reports. Founded in 1995 as a purely electronic journal, it has adapted its format to include a full-color print edition, and adjusted its aims and scope fit the fast-changing field of molecular modeling, with a particular focus on three-dimensional modeling.
Today, the journal covers all aspects of molecular modeling including life science modeling; materials modeling; new methods; and computational chemistry.
Topics include computer-aided molecular design; rational drug design, de novo ligand design, receptor modeling and docking; cheminformatics, data analysis, visualization and mining; computational medicinal chemistry; homology modeling; simulation of peptides, DNA and other biopolymers; quantitative structure-activity relationships (QSAR) and ADME-modeling; modeling of biological reaction mechanisms; and combined experimental and computational studies in which calculations play a major role.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


