{"title":"Genotype Distribution and Migration Patterns of Hepatitis C Virus in Shandong Province, China: Molecular Epidemiology and Phylogenetic Study.","authors":"Lin Lin, Guoyong Wang, Lianzheng Hao, Tingbin Yan","doi":"10.2196/60207","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>Hepatitis C virus (HCV) remains a significant public health concern in China, particularly in Shandong Province, where detailed molecular epidemiological data are limited. HCV exhibits substantial genetic diversity, and understanding its genotype distribution and transmission dynamics is critical for developing effective control strategies.</p><p><strong>Objective: </strong>This study aimed to investigate the genetic diversity, geographic dissemination, and evolutionary history of HCV genotypes in Shandong Province, China, using molecular techniques and phylogenetic methods.</p><p><strong>Methods: </strong>A total of 320 HCV-positive serum samples were collected from multiple hospitals across Shandong Province between 2013 and 2021. HCV RNA was extracted and amplified targeting the 5' untranslated region (UTR), Core, and NS5B regions. Sequencing was conducted, and genotypes were determined using the National Center for Biotechnology Information's Basic Local Alignment Search Tool (NCBI BLAST). Phylogenetic trees were constructed using maximum likelihood methods with the general time reversible with Gamma-distributed rate variation among sites [(GTR)+Gamma model]. The temporal and geographic evolution of the major subtypes (1b and 2a) was analyzed using Bayesian Markov chain Monte Carlo (MCMC) methods implemented in Bayesian Evolutionary Analysis Sampling Trees (BEAST). The Bayesian skyline plot (BSP) was used to infer population dynamics and estimate the time to the most recent common ancestor (tMRCA).</p><p><strong>Results: </strong>Genotypes 1b (n=165) and 2a (n=131) were identified as the predominant subtypes, with a small number of genotypes 3b, 6a, 6k, and potential recombinant strains also detected. Phylogenetic analysis revealed distinct evolutionary clustering of 1b and 2a strains, suggesting multiple diffusion events within the province. The tMRCA of subtypes 1b and 2a were estimated to be 1957 and 1979, respectively. Bayesian skyline analysis showed that both subtypes experienced long-term population stability, followed by a rapid expansion period between 2014 and 2019 (1b) and 2014 to 2016 (2a), respectively. The analysis also identified key transmission hubs such as Jinan, Liaocheng, Tai'an, and Dezhou, indicating city-level variations in HCV spread.</p><p><strong>Conclusions: </strong>This study provides data-supported insights into the genotypic landscape and evolutionary patterns of HCV in Shandong Province. The identification of dominant subtypes, potential recombinant strains, and regional transmission pathways enhances our understanding of local HCV epidemiology. These findings have implications for public health policy, resource allocation, and targeted treatment strategies. The integration of molecular epidemiology and phylogenetics offers a valuable model for infectious disease surveillance and control in similar settings.</p>","PeriodicalId":56334,"journal":{"name":"JMIR Medical Informatics","volume":"13 ","pages":"e60207"},"PeriodicalIF":3.8000,"publicationDate":"2025-08-18","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12360734/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"JMIR Medical Informatics","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.2196/60207","RegionNum":3,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"MEDICAL INFORMATICS","Score":null,"Total":0}
引用次数: 0
Abstract
Background: Hepatitis C virus (HCV) remains a significant public health concern in China, particularly in Shandong Province, where detailed molecular epidemiological data are limited. HCV exhibits substantial genetic diversity, and understanding its genotype distribution and transmission dynamics is critical for developing effective control strategies.
Objective: This study aimed to investigate the genetic diversity, geographic dissemination, and evolutionary history of HCV genotypes in Shandong Province, China, using molecular techniques and phylogenetic methods.
Methods: A total of 320 HCV-positive serum samples were collected from multiple hospitals across Shandong Province between 2013 and 2021. HCV RNA was extracted and amplified targeting the 5' untranslated region (UTR), Core, and NS5B regions. Sequencing was conducted, and genotypes were determined using the National Center for Biotechnology Information's Basic Local Alignment Search Tool (NCBI BLAST). Phylogenetic trees were constructed using maximum likelihood methods with the general time reversible with Gamma-distributed rate variation among sites [(GTR)+Gamma model]. The temporal and geographic evolution of the major subtypes (1b and 2a) was analyzed using Bayesian Markov chain Monte Carlo (MCMC) methods implemented in Bayesian Evolutionary Analysis Sampling Trees (BEAST). The Bayesian skyline plot (BSP) was used to infer population dynamics and estimate the time to the most recent common ancestor (tMRCA).
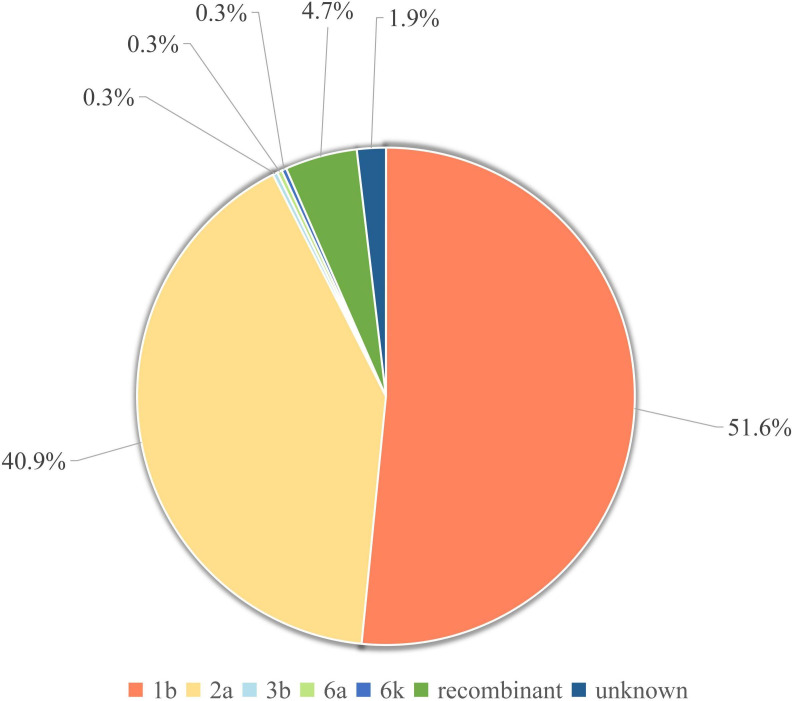
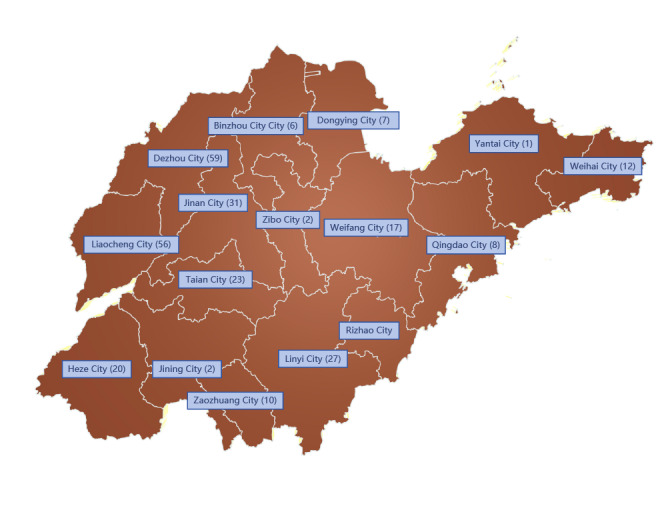
Results: Genotypes 1b (n=165) and 2a (n=131) were identified as the predominant subtypes, with a small number of genotypes 3b, 6a, 6k, and potential recombinant strains also detected. Phylogenetic analysis revealed distinct evolutionary clustering of 1b and 2a strains, suggesting multiple diffusion events within the province. The tMRCA of subtypes 1b and 2a were estimated to be 1957 and 1979, respectively. Bayesian skyline analysis showed that both subtypes experienced long-term population stability, followed by a rapid expansion period between 2014 and 2019 (1b) and 2014 to 2016 (2a), respectively. The analysis also identified key transmission hubs such as Jinan, Liaocheng, Tai'an, and Dezhou, indicating city-level variations in HCV spread.
Conclusions: This study provides data-supported insights into the genotypic landscape and evolutionary patterns of HCV in Shandong Province. The identification of dominant subtypes, potential recombinant strains, and regional transmission pathways enhances our understanding of local HCV epidemiology. These findings have implications for public health policy, resource allocation, and targeted treatment strategies. The integration of molecular epidemiology and phylogenetics offers a valuable model for infectious disease surveillance and control in similar settings.
期刊介绍:
JMIR Medical Informatics (JMI, ISSN 2291-9694) is a top-rated, tier A journal which focuses on clinical informatics, big data in health and health care, decision support for health professionals, electronic health records, ehealth infrastructures and implementation. It has a focus on applied, translational research, with a broad readership including clinicians, CIOs, engineers, industry and health informatics professionals.
Published by JMIR Publications, publisher of the Journal of Medical Internet Research (JMIR), the leading eHealth/mHealth journal (Impact Factor 2016: 5.175), JMIR Med Inform has a slightly different scope (emphasizing more on applications for clinicians and health professionals rather than consumers/citizens, which is the focus of JMIR), publishes even faster, and also allows papers which are more technical or more formative than what would be published in the Journal of Medical Internet Research.


 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


