{"title":"Cross-cohort microbiome-wide study reveals consistent alterations in the gut bacteriome, but not the gut mycobiome, in patients with hypertension.","authors":"Yidan Gao, Dangrang Wang, Tong Lu, Kan Liu, Shenghui Li, Jian Kang, Shanshan Sha, Guorui Xing, Lin Cheng, Shao Fan, Wei Yang, Qiulong Yan, Yanchun Ding, Dafeng Xu","doi":"10.1128/msystems.00657-25","DOIUrl":null,"url":null,"abstract":"<p><p>Hypertension, one of the most prevalent cardiovascular diseases, has been linked to the gut microbiota. However, there is a lack of well-defined, cross-population validated gut microbial signatures associated with hypertension, particularly at both the bacterial and fungal levels. To address this gap, we conducted a metagenome-wide analysis of fecal samples from 159 hypertensive patients and 101 healthy controls, using two publicly available data sets from the Beijing and Dalian regions. Our results showed that hypertensive patients exhibit a significant reduction in gut bacterial diversity, accompanied by substantial alterations in bacterial composition. A total of 61 bacterial species were identified with significantly different relative abundance between patients and controls across both regions (combined <i>P</i> < 0.05, <i>q</i> = 0.25). Hypertension-enriched species included <i>Lachnospiraceae</i> (<i>Clostridium symbiosum</i>, <i>Enterocloster bolteae</i>) and <i>Clostridium</i> sp. AT4, while <i>Lachnospiraceae bacterium</i>, <i>Firmicutes bacterium</i>, and <i>Clostridium</i> sp. AM49 4BH were significantly decreased in hypertensive patients. In contrast, no significant differences were observed in gut fungal diversity between hypertensive patients and healthy controls, and only minor differences in fungal composition were noted. Specifically, six fungal species were identified as potentially significant in the combined data set (<i>P</i> < 0.05, <i>q</i> = 0.73), but they only <i>Blastomyces emzantsi</i> c231 meet the same consistency across the two cohorts as the bacterial signatures. Furthermore, we developed classification models using gut bacterial and fungal signatures to distinguish hypertension patients from healthy controls. The bacterium-based models achieved area under the curves (AUCs) greater than 0.70 in cross-cohort classification and validation, while the fungus-based models only achieved AUCs between 0.55 and 0.57. In summary, our study identifies cross-cohort gut bacterial and fungal signatures associated with hypertension, suggesting that the gut bacteriome may serve as a more reliable target for hypertension intervention compared to the gut mycobiome.</p><p><strong>Importance: </strong>Hypertension (HTN) represents a global health burden affecting billions of individuals worldwide; however, the relationship between HTN and gut microbial ecosystems remains inadequately characterized. This study presents the first cross-cohort microbiome analysis revealing significant alterations in the gut bacteriome of HTN patients, with limited changes observed in the mycobiome. These findings highlight the critical role of the gut bacteriome in the pathogenesis of HTN and provide new microbial biomarkers for early diagnosis. Furthermore, the identification of bacterial species establishes a foundation for future intervention approaches, enhancing the applicability of microbiome research in cardiovascular health and opening new avenues for related studies in this field.</p>","PeriodicalId":18819,"journal":{"name":"mSystems","volume":" ","pages":"e0065725"},"PeriodicalIF":4.6000,"publicationDate":"2025-09-23","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12455987/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"mSystems","FirstCategoryId":"99","ListUrlMain":"https://doi.org/10.1128/msystems.00657-25","RegionNum":2,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2025/8/15 0:00:00","PubModel":"Epub","JCR":"Q1","JCRName":"MICROBIOLOGY","Score":null,"Total":0}
引用次数: 0
Abstract
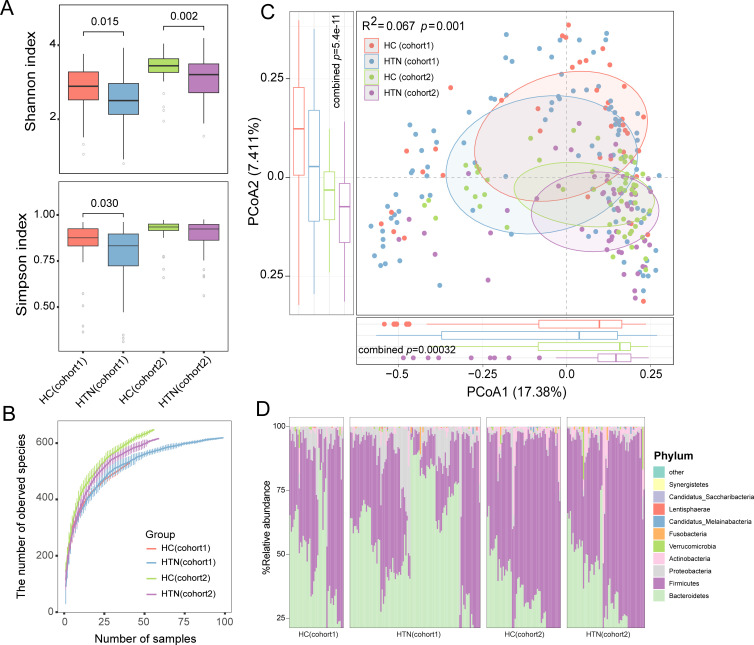
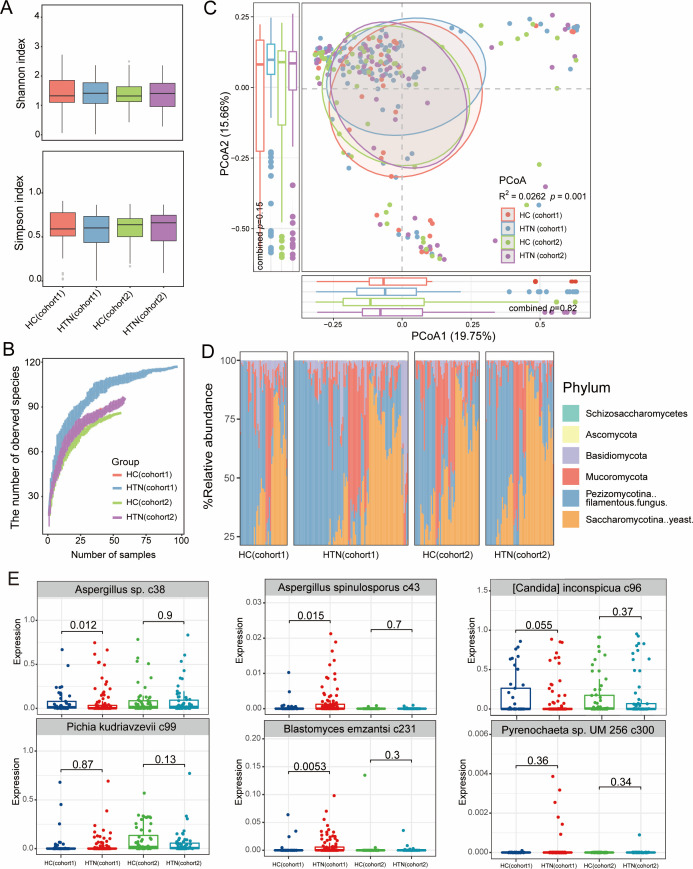
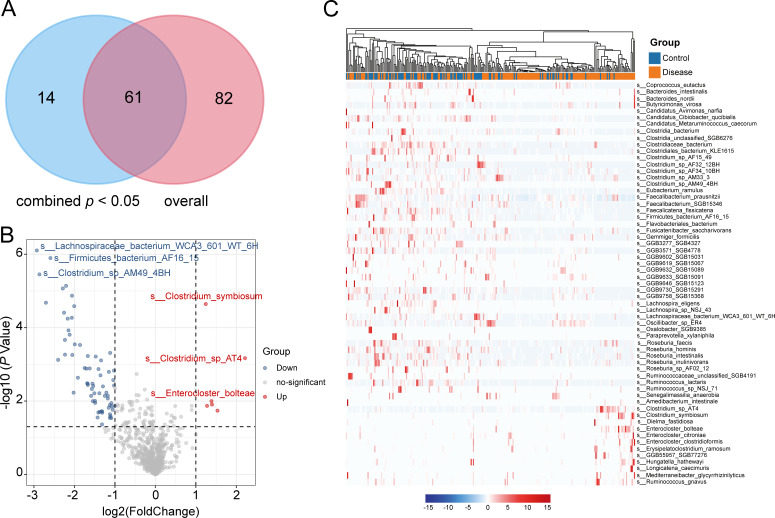
Hypertension, one of the most prevalent cardiovascular diseases, has been linked to the gut microbiota. However, there is a lack of well-defined, cross-population validated gut microbial signatures associated with hypertension, particularly at both the bacterial and fungal levels. To address this gap, we conducted a metagenome-wide analysis of fecal samples from 159 hypertensive patients and 101 healthy controls, using two publicly available data sets from the Beijing and Dalian regions. Our results showed that hypertensive patients exhibit a significant reduction in gut bacterial diversity, accompanied by substantial alterations in bacterial composition. A total of 61 bacterial species were identified with significantly different relative abundance between patients and controls across both regions (combined P < 0.05, q = 0.25). Hypertension-enriched species included Lachnospiraceae (Clostridium symbiosum, Enterocloster bolteae) and Clostridium sp. AT4, while Lachnospiraceae bacterium, Firmicutes bacterium, and Clostridium sp. AM49 4BH were significantly decreased in hypertensive patients. In contrast, no significant differences were observed in gut fungal diversity between hypertensive patients and healthy controls, and only minor differences in fungal composition were noted. Specifically, six fungal species were identified as potentially significant in the combined data set (P < 0.05, q = 0.73), but they only Blastomyces emzantsi c231 meet the same consistency across the two cohorts as the bacterial signatures. Furthermore, we developed classification models using gut bacterial and fungal signatures to distinguish hypertension patients from healthy controls. The bacterium-based models achieved area under the curves (AUCs) greater than 0.70 in cross-cohort classification and validation, while the fungus-based models only achieved AUCs between 0.55 and 0.57. In summary, our study identifies cross-cohort gut bacterial and fungal signatures associated with hypertension, suggesting that the gut bacteriome may serve as a more reliable target for hypertension intervention compared to the gut mycobiome.
Importance: Hypertension (HTN) represents a global health burden affecting billions of individuals worldwide; however, the relationship between HTN and gut microbial ecosystems remains inadequately characterized. This study presents the first cross-cohort microbiome analysis revealing significant alterations in the gut bacteriome of HTN patients, with limited changes observed in the mycobiome. These findings highlight the critical role of the gut bacteriome in the pathogenesis of HTN and provide new microbial biomarkers for early diagnosis. Furthermore, the identification of bacterial species establishes a foundation for future intervention approaches, enhancing the applicability of microbiome research in cardiovascular health and opening new avenues for related studies in this field.
mSystemsBiochemistry, Genetics and Molecular Biology-Biochemistry
CiteScore
10.50
自引率
3.10%
发文量
308
审稿时长
13 weeks
期刊介绍:
mSystems™ will publish preeminent work that stems from applying technologies for high-throughput analyses to achieve insights into the metabolic and regulatory systems at the scale of both the single cell and microbial communities. The scope of mSystems™ encompasses all important biological and biochemical findings drawn from analyses of large data sets, as well as new computational approaches for deriving these insights. mSystems™ will welcome submissions from researchers who focus on the microbiome, genomics, metagenomics, transcriptomics, metabolomics, proteomics, glycomics, bioinformatics, and computational microbiology. mSystems™ will provide streamlined decisions, while carrying on ASM''s tradition of rigorous peer review.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


