A unified graph-based approach for protein function prediction using AlphaFold structures and sequence features
IF 3.1
4区 生物学
Q2 BIOLOGY
引用次数: 0
Abstract
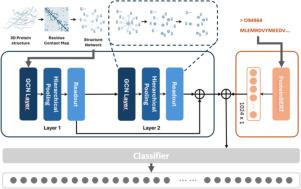
Predicting protein function is a key challenge in computational biology with broad implications for understanding biological systems and disease mechanisms. Traditional deep learning approaches rely heavily on protein sequence data and protein–protein interaction (PPI) networks, often neglecting structural information due to limited availability of experimentally resolved protein structures. The advent of AlphaFold, which predicts protein structures with near-atomic accuracy, provides an opportunity to integrate structural context into function prediction. In this study, we propose StructSeq2GO, a novel hybrid model that combines structural and sequence information. StructSeq2GO employs graph representation learning to extract structural features from AlphaFold-predicted protein structures and integrates them with sequence embeddings derived from the ProteinBERT language model to predict Gene Ontology (GO) labels. Experimental evaluations demonstrate that StructSeq2GO achieves state-of-the-art performance across three GO domains, with scores of 0.485, 0.681, and 0.663, AUC scores of 0.764, 0.939, and 0.891, and AUPR scores of 0.688, 0.763, and 0.702 for the Biological Process (BPO), Cellular Component (CCO), and Molecular Function (MFO) ontologies, respectively. These results highlight the critical importance of structural information and the efficacy of ProteinBERT in enhancing protein function prediction, as structure provides spatial and biochemical context not captured by sequence alone. The model’s performance is influenced by the quality of AlphaFold structural predictions and may benefit from future improvements in structure confidence modeling. Additionally, extending StructSeq2GO to predict pathway-level or disease-related annotations could broaden its biological utility.
使用AlphaFold结构和序列特征的统一的基于图的蛋白质功能预测方法
预测蛋白质功能是计算生物学中的一个关键挑战,对理解生物系统和疾病机制具有广泛的意义。传统的深度学习方法严重依赖于蛋白质序列数据和蛋白质相互作用(PPI)网络,由于实验解决蛋白质结构的可用性有限,通常忽略了结构信息。AlphaFold以接近原子的精度预测蛋白质结构,为将结构背景整合到功能预测中提供了机会。在这项研究中,我们提出了StructSeq2GO,一种结合结构和序列信息的新型混合模型。StructSeq2GO利用图表示学习从alphafold预测的蛋白质结构中提取结构特征,并将其与ProteinBERT语言模型衍生的序列嵌入相结合,预测基因本体(Gene Ontology, GO)标签。实验评估表明,StructSeq2GO在三个GO域上达到了最先进的性能,生物过程(BPO)、细胞成分(CCO)和分子功能(MFO)本体的Fmax得分分别为0.485、0.681和0.663,AUC得分分别为0.764、0.939和0.891,AUPR得分分别为0.688、0.763和0.702。这些结果强调了结构信息和ProteinBERT在增强蛋白质功能预测方面的重要性,因为结构提供了空间和生化背景,而不是单独由序列捕获。该模型的性能受到AlphaFold结构预测质量的影响,并可能受益于未来结构置信度建模的改进。此外,扩展StructSeq2GO来预测通路水平或疾病相关的注释可以拓宽其生物学用途。
本文章由计算机程序翻译,如有差异,请以英文原文为准。
求助全文
约1分钟内获得全文
求助全文
来源期刊

Computational Biology and Chemistry
生物-计算机:跨学科应用
CiteScore
6.10
自引率
3.20%
发文量
142
审稿时长
24 days
期刊介绍:
Computational Biology and Chemistry publishes original research papers and review articles in all areas of computational life sciences. High quality research contributions with a major computational component in the areas of nucleic acid and protein sequence research, molecular evolution, molecular genetics (functional genomics and proteomics), theory and practice of either biology-specific or chemical-biology-specific modeling, and structural biology of nucleic acids and proteins are particularly welcome. Exceptionally high quality research work in bioinformatics, systems biology, ecology, computational pharmacology, metabolism, biomedical engineering, epidemiology, and statistical genetics will also be considered.
Given their inherent uncertainty, protein modeling and molecular docking studies should be thoroughly validated. In the absence of experimental results for validation, the use of molecular dynamics simulations along with detailed free energy calculations, for example, should be used as complementary techniques to support the major conclusions. Submissions of premature modeling exercises without additional biological insights will not be considered.
Review articles will generally be commissioned by the editors and should not be submitted to the journal without explicit invitation. However prospective authors are welcome to send a brief (one to three pages) synopsis, which will be evaluated by the editors.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


