S K Nemani, X Xiao, S Notari, I Cali, K Lundberg, L Cracco, B S Appleby, W K Surewicz, V L Sim, P Gambetti
{"title":"Variably protease-sensitive prionopathy: mass spectrometry analysis of the pathogenic prion protein provides a new perspective.","authors":"S K Nemani, X Xiao, S Notari, I Cali, K Lundberg, L Cracco, B S Appleby, W K Surewicz, V L Sim, P Gambetti","doi":"10.1186/s40478-025-02079-9","DOIUrl":null,"url":null,"abstract":"<p><p>Variably protease-sensitive prionopathy (VPSPr) is a rare and complex prion disease that differs from sporadic Creutzfeldt-Jakob disease (sCJD) in its clinical and histopathological phenotypes. VPSPr also features a variety of fragments generated by the disease-causing prion protein (PrP<sup>D</sup>). However, accurately determining the number and sequence of these fragments has been challenging when relying solely on epitope mapping with existing antibodies. To address these challenges, we performed mass spectrometry analyses and designed epitope mapping experiments to determine the primary structure and verify the presence or absence of the anchor in the VPSPr proteinase K-resistant and deglycosylated PrP<sup>D</sup> fragments. All three N-terminus fragments, with reported molecular weights of 20, 17, and 7 kDa, likely share Ser97 as the N-terminal amino acid. The C-terminus of the internal 7 kDa fragment is ragged, extending from Phe141 to Met154, while the 20 kDa and 17 kDa fragments differ only in the absence of the anchor in the latter. The three fragments belonging to the C-terminus group have previously been reported to have electrophoretic mobilities of 18, 12/13, and 8-9 kDa. After deglycosylation, the 18 kDa fragment was not detected. The 12 kDa component of the 12/13 kDa fragment was found to have a ragged N-terminus between Tyr162 and Asp181 and the anchor, while the 8 kDa fragment represented the anchorless version of the 12 kDa fragment. Unexpectedly, a second approximately 8 kDa fragment was identified that bore the anchor but had a shorter, ragged N-terminus ranging from Gly195 to Phe198. Calculation based on sequencing data revealed that the actual molecular masses of the 20, 17, and 12 kDa fragments are 1-2 kDa lighter. Moreover, the primary structures of the 20, 17, and 12 kDa fragments match those of the 19, 17, and 12 kDa fragments associated with sCJD type 2. Our findings provide new insights into the characteristics of the deglycosylated, PK-resistant fragments in VPSPr, which will likely assist in interpreting future high-resolution studies of amyloid fibrils in this disease.</p>","PeriodicalId":6914,"journal":{"name":"Acta Neuropathologica Communications","volume":"13 1","pages":"172"},"PeriodicalIF":5.7000,"publicationDate":"2025-08-12","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12345132/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Acta Neuropathologica Communications","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1186/s40478-025-02079-9","RegionNum":2,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"NEUROSCIENCES","Score":null,"Total":0}
引用次数: 0
Abstract
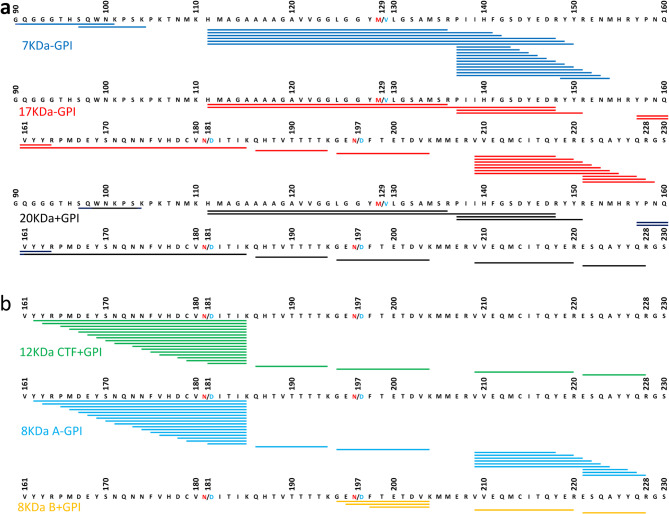
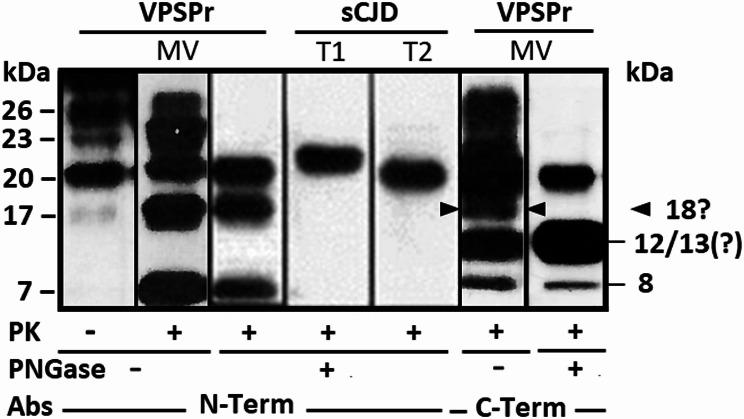
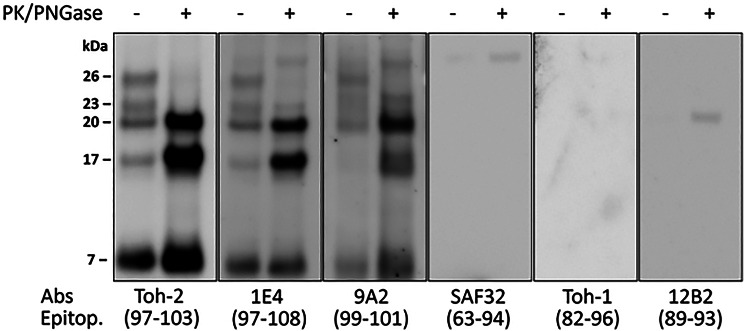
Variably protease-sensitive prionopathy (VPSPr) is a rare and complex prion disease that differs from sporadic Creutzfeldt-Jakob disease (sCJD) in its clinical and histopathological phenotypes. VPSPr also features a variety of fragments generated by the disease-causing prion protein (PrPD). However, accurately determining the number and sequence of these fragments has been challenging when relying solely on epitope mapping with existing antibodies. To address these challenges, we performed mass spectrometry analyses and designed epitope mapping experiments to determine the primary structure and verify the presence or absence of the anchor in the VPSPr proteinase K-resistant and deglycosylated PrPD fragments. All three N-terminus fragments, with reported molecular weights of 20, 17, and 7 kDa, likely share Ser97 as the N-terminal amino acid. The C-terminus of the internal 7 kDa fragment is ragged, extending from Phe141 to Met154, while the 20 kDa and 17 kDa fragments differ only in the absence of the anchor in the latter. The three fragments belonging to the C-terminus group have previously been reported to have electrophoretic mobilities of 18, 12/13, and 8-9 kDa. After deglycosylation, the 18 kDa fragment was not detected. The 12 kDa component of the 12/13 kDa fragment was found to have a ragged N-terminus between Tyr162 and Asp181 and the anchor, while the 8 kDa fragment represented the anchorless version of the 12 kDa fragment. Unexpectedly, a second approximately 8 kDa fragment was identified that bore the anchor but had a shorter, ragged N-terminus ranging from Gly195 to Phe198. Calculation based on sequencing data revealed that the actual molecular masses of the 20, 17, and 12 kDa fragments are 1-2 kDa lighter. Moreover, the primary structures of the 20, 17, and 12 kDa fragments match those of the 19, 17, and 12 kDa fragments associated with sCJD type 2. Our findings provide new insights into the characteristics of the deglycosylated, PK-resistant fragments in VPSPr, which will likely assist in interpreting future high-resolution studies of amyloid fibrils in this disease.
期刊介绍:
"Acta Neuropathologica Communications (ANC)" is a peer-reviewed journal that specializes in the rapid publication of research articles focused on the mechanisms underlying neurological diseases. The journal emphasizes the use of molecular, cellular, and morphological techniques applied to experimental or human tissues to investigate the pathogenesis of neurological disorders.
ANC is committed to a fast-track publication process, aiming to publish accepted manuscripts within two months of submission. This expedited timeline is designed to ensure that the latest findings in neuroscience and pathology are disseminated quickly to the scientific community, fostering rapid advancements in the field of neurology and neuroscience. The journal's focus on cutting-edge research and its swift publication schedule make it a valuable resource for researchers, clinicians, and other professionals interested in the study and treatment of neurological conditions.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


