Nelson R. C. Junior, Maicon Pierre Lourenço, Breno R. L. Galvão
{"title":"A genetic algorithm search for the global minima of sodium nanoclusters using a tailored DFTB approach","authors":"Nelson R. C. Junior, Maicon Pierre Lourenço, Breno R. L. Galvão","doi":"10.1007/s00894-025-06457-x","DOIUrl":null,"url":null,"abstract":"<div><h3>Context</h3><p>Obtaining the global minimum in the potential energy hypersurface of nanoclusters is a very difficult task, due to the large number of degrees of freedom and the vast number of local minima. However, the discovery of such minima provides the geometrical arrangement that is more likely to occur, which is a key step in computing the properties of such particles. Here we developed a genetic algorithm (GA) including a gradient adjustment in each local optimization, to obtain an efficient GA, which is particularly useful when the algorithm is coupled with electronic structure methods. The idea is first validated, and then used to predict the minima of large sodium nanoclusters up to one hundred atoms.</p><h3>Methods</h3><p>To validate the algorithm, we analyzed its efficiency in obtaining the global minima of Lennard–Jones clusters, whose solutions are well known and can be used as benchmark. The new GA is compared to a random search and a standard GA. For exploring the potential energy surface of sodium clusters, we employ the Density-Functional Tight-Binding (DFTB) method, with parameters that have been tuned specifically to such clusters, thus enhancing its reliability for this specific application.</p></div>","PeriodicalId":651,"journal":{"name":"Journal of Molecular Modeling","volume":"31 9","pages":""},"PeriodicalIF":2.5000,"publicationDate":"2025-08-13","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Molecular Modeling","FirstCategoryId":"92","ListUrlMain":"https://link.springer.com/article/10.1007/s00894-025-06457-x","RegionNum":4,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q4","JCRName":"BIOCHEMISTRY & MOLECULAR BIOLOGY","Score":null,"Total":0}
引用次数: 0
Abstract
Context
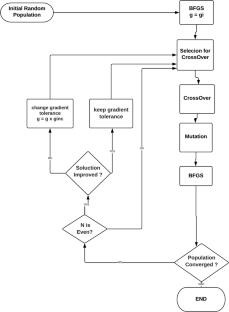
Obtaining the global minimum in the potential energy hypersurface of nanoclusters is a very difficult task, due to the large number of degrees of freedom and the vast number of local minima. However, the discovery of such minima provides the geometrical arrangement that is more likely to occur, which is a key step in computing the properties of such particles. Here we developed a genetic algorithm (GA) including a gradient adjustment in each local optimization, to obtain an efficient GA, which is particularly useful when the algorithm is coupled with electronic structure methods. The idea is first validated, and then used to predict the minima of large sodium nanoclusters up to one hundred atoms.
Methods
To validate the algorithm, we analyzed its efficiency in obtaining the global minima of Lennard–Jones clusters, whose solutions are well known and can be used as benchmark. The new GA is compared to a random search and a standard GA. For exploring the potential energy surface of sodium clusters, we employ the Density-Functional Tight-Binding (DFTB) method, with parameters that have been tuned specifically to such clusters, thus enhancing its reliability for this specific application.
期刊介绍:
The Journal of Molecular Modeling focuses on "hardcore" modeling, publishing high-quality research and reports. Founded in 1995 as a purely electronic journal, it has adapted its format to include a full-color print edition, and adjusted its aims and scope fit the fast-changing field of molecular modeling, with a particular focus on three-dimensional modeling.
Today, the journal covers all aspects of molecular modeling including life science modeling; materials modeling; new methods; and computational chemistry.
Topics include computer-aided molecular design; rational drug design, de novo ligand design, receptor modeling and docking; cheminformatics, data analysis, visualization and mining; computational medicinal chemistry; homology modeling; simulation of peptides, DNA and other biopolymers; quantitative structure-activity relationships (QSAR) and ADME-modeling; modeling of biological reaction mechanisms; and combined experimental and computational studies in which calculations play a major role.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


