Anshita Goel, Benjamin J Tura, Joanne D Stockton, Nicholas Tovey, Luke Ames, Andrew D Beggs, Maurice P Zeegers, Nicholas D James, K K Cheng, Richard T Bryan, Douglas G Ward, Roland Arnold
{"title":"Detection of genome-wide methylation changes in bladder cancer by long-read sequencing of urinary DNA.","authors":"Anshita Goel, Benjamin J Tura, Joanne D Stockton, Nicholas Tovey, Luke Ames, Andrew D Beggs, Maurice P Zeegers, Nicholas D James, K K Cheng, Richard T Bryan, Douglas G Ward, Roland Arnold","doi":"10.1186/s13148-025-01946-5","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>Non-invasive urine tests for bladder cancer (BC) could reduce dependence on flexible cystoscopy for diagnosis and surveillance. Most recent developments in urine testing are based on targeted detection of genomic and/or epigenomic markers. We hypothesised that long-read whole-genome sequencing of urinary DNA with direct methylation profiling may allow accurate BC detection and insights into disease biology. However, the feasibility of such an approach has not yet been reported.</p><p><strong>Methods: </strong>We applied long-read whole-genome sequencing with direct methylation detection to urine cell pellet DNA (ucpDNA) from 21 haematuria clinic patients: 13 BCs and 8 non-BCs. The modkit Hidden Markov Model algorithm was used to define differentially methylated regions across the genome. The ability to discriminate between BC and non-BC, and the cellular pathways affected were tested using PCA, h-clust and GSEA.</p><p><strong>Results: </strong>We observed global hypomethylation and cancer-specific patterns of promoter hypermethylation in urine from BC patients. Sequencing of a single ucpDNA sample per flow cell yielded read depths of 18-34x; furthermore, BC methylation patterns were also evident with 2-5x multiplex sequencing. Copy number changes were also evident in ucpDNAs from BC patients. A limitation of the study is the small number of samples analysed; however, the detection of cancer-specific events demonstrates the feasibility of the approach, both in single and multiplexed flow-cell runs.</p><p><strong>Conclusions: </strong>Even at low-read depths, genome-wide methylation patterns in urinary DNA reflect the presence of BC, potentially permitting rapid, non-invasive and cost-effective BC detection.</p>","PeriodicalId":10366,"journal":{"name":"Clinical Epigenetics","volume":"17 1","pages":"141"},"PeriodicalIF":4.4000,"publicationDate":"2025-08-11","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12337379/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Clinical Epigenetics","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1186/s13148-025-01946-5","RegionNum":2,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"GENETICS & HEREDITY","Score":null,"Total":0}
引用次数: 0
Abstract
Background: Non-invasive urine tests for bladder cancer (BC) could reduce dependence on flexible cystoscopy for diagnosis and surveillance. Most recent developments in urine testing are based on targeted detection of genomic and/or epigenomic markers. We hypothesised that long-read whole-genome sequencing of urinary DNA with direct methylation profiling may allow accurate BC detection and insights into disease biology. However, the feasibility of such an approach has not yet been reported.
Methods: We applied long-read whole-genome sequencing with direct methylation detection to urine cell pellet DNA (ucpDNA) from 21 haematuria clinic patients: 13 BCs and 8 non-BCs. The modkit Hidden Markov Model algorithm was used to define differentially methylated regions across the genome. The ability to discriminate between BC and non-BC, and the cellular pathways affected were tested using PCA, h-clust and GSEA.
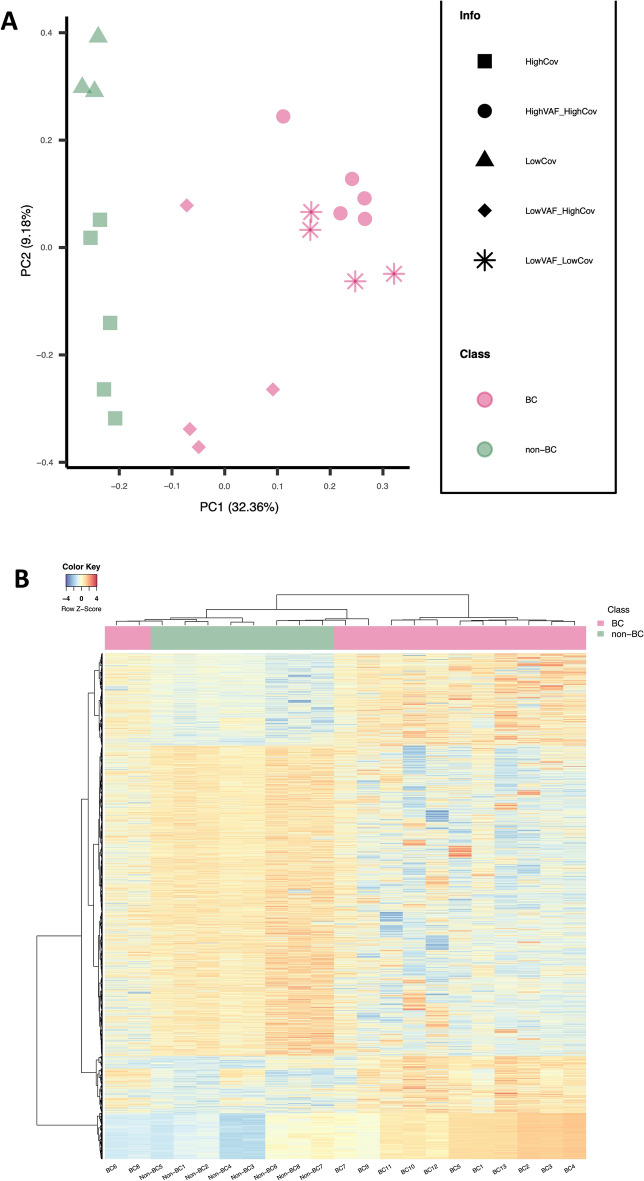
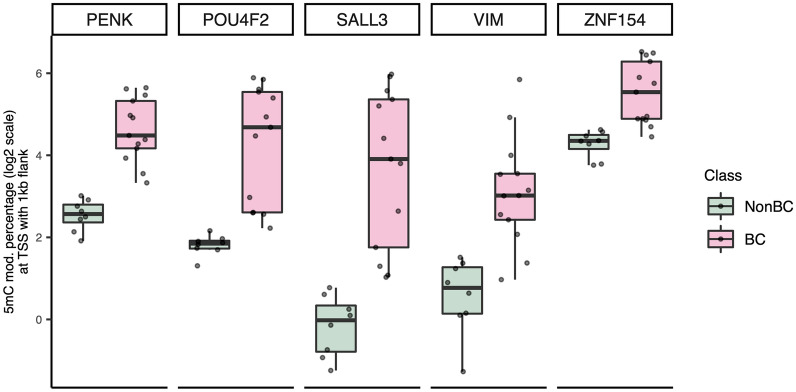
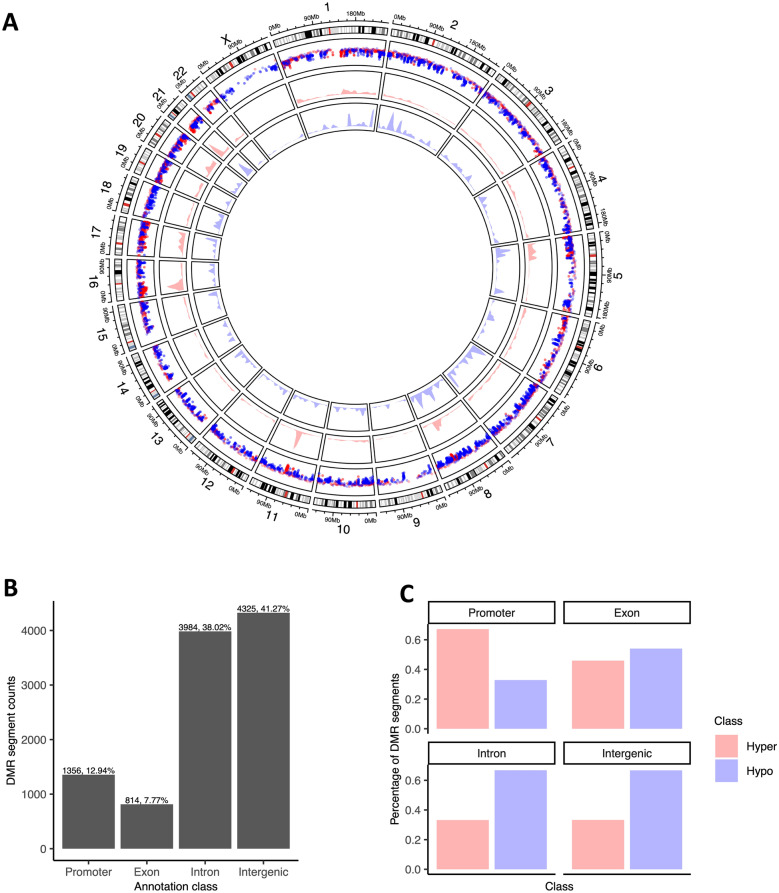
Results: We observed global hypomethylation and cancer-specific patterns of promoter hypermethylation in urine from BC patients. Sequencing of a single ucpDNA sample per flow cell yielded read depths of 18-34x; furthermore, BC methylation patterns were also evident with 2-5x multiplex sequencing. Copy number changes were also evident in ucpDNAs from BC patients. A limitation of the study is the small number of samples analysed; however, the detection of cancer-specific events demonstrates the feasibility of the approach, both in single and multiplexed flow-cell runs.
Conclusions: Even at low-read depths, genome-wide methylation patterns in urinary DNA reflect the presence of BC, potentially permitting rapid, non-invasive and cost-effective BC detection.
期刊介绍:
Clinical Epigenetics, the official journal of the Clinical Epigenetics Society, is an open access, peer-reviewed journal that encompasses all aspects of epigenetic principles and mechanisms in relation to human disease, diagnosis and therapy. Clinical trials and research in disease model organisms are particularly welcome.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


