Vitória S. Reis, Luciana Guimarães, Clebio S. Nascimento Jr.
{"title":"A computational study on the enantioselective separation of cyhalothrin enantiomers by β-cyclodextrins","authors":"Vitória S. Reis, Luciana Guimarães, Clebio S. Nascimento Jr.","doi":"10.1007/s00894-025-06469-7","DOIUrl":null,"url":null,"abstract":"<div><h3>Context</h3><p>This computational study investigates the enantioselective separation of λ-cyhalothrin (CLT) enantiomers, ( +)-[S]-CLT and ( −)-[R]-CLT, using native β-cyclodextrin (β-CD) and its sulfated derivative (S-β-CD) as chiral selectors. Our calculations, employing B97D/6-31G(d,p)//PM3 and B97X-D3/6-31G(d,p)//GFN2-xTB methodologies, consistently demonstrate that the ( −)-[R]-CLT enantiomer forms more stable inclusion complexes with both selectors, predicting a longer migration time compared to ( +)-[S]-CLT. This enhanced stability is primarily due to favorable hydrophobic interactions, with additional hydrogen bonding contributing in the case of S-β-CD. S-β-CD exhibited superior separation efficiency, as evidenced by higher ΔΔ<i>G</i> values, likely due to structural modifications induced by sulfate groups that optimize steric fitting and van der Waals interactions within the cyclodextrin cavity. These findings highlight the potential of S-β-CD for efficient enantiomeric separation of CLT and underscore the utility of computational chemistry in understanding chiral recognition mechanisms.</p><h3>Methods</h3><p>Two distinct theoretical methodologies, B97D/6-31G(d,p)//PM3 and ωB97X-D3/6-31G(d,p)//GFN2-xTB, were employed to elucidate the structural and energetic properties of these diastereomeric complexes. The calculations were performed on Gaussian 09 and ORCA 5.0 software packages.\n</p></div>","PeriodicalId":651,"journal":{"name":"Journal of Molecular Modeling","volume":"31 9","pages":""},"PeriodicalIF":2.5000,"publicationDate":"2025-08-11","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Molecular Modeling","FirstCategoryId":"92","ListUrlMain":"https://link.springer.com/article/10.1007/s00894-025-06469-7","RegionNum":4,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q4","JCRName":"BIOCHEMISTRY & MOLECULAR BIOLOGY","Score":null,"Total":0}
引用次数: 0
Abstract
Context
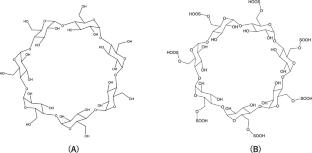
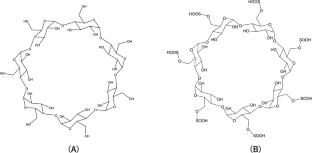
This computational study investigates the enantioselective separation of λ-cyhalothrin (CLT) enantiomers, ( +)-[S]-CLT and ( −)-[R]-CLT, using native β-cyclodextrin (β-CD) and its sulfated derivative (S-β-CD) as chiral selectors. Our calculations, employing B97D/6-31G(d,p)//PM3 and B97X-D3/6-31G(d,p)//GFN2-xTB methodologies, consistently demonstrate that the ( −)-[R]-CLT enantiomer forms more stable inclusion complexes with both selectors, predicting a longer migration time compared to ( +)-[S]-CLT. This enhanced stability is primarily due to favorable hydrophobic interactions, with additional hydrogen bonding contributing in the case of S-β-CD. S-β-CD exhibited superior separation efficiency, as evidenced by higher ΔΔG values, likely due to structural modifications induced by sulfate groups that optimize steric fitting and van der Waals interactions within the cyclodextrin cavity. These findings highlight the potential of S-β-CD for efficient enantiomeric separation of CLT and underscore the utility of computational chemistry in understanding chiral recognition mechanisms.
Methods
Two distinct theoretical methodologies, B97D/6-31G(d,p)//PM3 and ωB97X-D3/6-31G(d,p)//GFN2-xTB, were employed to elucidate the structural and energetic properties of these diastereomeric complexes. The calculations were performed on Gaussian 09 and ORCA 5.0 software packages.
期刊介绍:
The Journal of Molecular Modeling focuses on "hardcore" modeling, publishing high-quality research and reports. Founded in 1995 as a purely electronic journal, it has adapted its format to include a full-color print edition, and adjusted its aims and scope fit the fast-changing field of molecular modeling, with a particular focus on three-dimensional modeling.
Today, the journal covers all aspects of molecular modeling including life science modeling; materials modeling; new methods; and computational chemistry.
Topics include computer-aided molecular design; rational drug design, de novo ligand design, receptor modeling and docking; cheminformatics, data analysis, visualization and mining; computational medicinal chemistry; homology modeling; simulation of peptides, DNA and other biopolymers; quantitative structure-activity relationships (QSAR) and ADME-modeling; modeling of biological reaction mechanisms; and combined experimental and computational studies in which calculations play a major role.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


