{"title":"Revisiting guidance on population sampling for highly polymorphic STR loci.","authors":"Sanne E Aalbers, Katherine B Gettings","doi":"10.1016/j.fsigen.2025.103336","DOIUrl":null,"url":null,"abstract":"<p><p>Population databases allow us to attach probabilities to DNA evidence by the estimation of genotype frequencies, which rely on accurate allele frequency estimates. As short tandem repeat (STR) marker sets for human identification have expanded to include more discriminating markers, and especially now that sequencing techniques allow us to distinguish between alleles based on variation in underlying base-pair structure, it is important to reevaluate existing guidance on population database sizes for the estimation of allele frequencies. In this paper, we revisit the topic of population sampling by focusing on the representation of alleles, i.e. whether alleles are observed or not, in a sample of individuals containing data for highly polymorphic autosomal STR loci. The effect of both length- and sequence-based STR data on population sample size implications are demonstrated, and differences between lesser and more polymorphic markers are discussed. The consequences of using a limited number of individuals are explored and the impact of increasing population sample sizes by combining different data sets is shown to help determine the point at which further sampling may no longer provide significant value. Finally, different approaches for accommodating previously unobserved alleles and their impact on DNA evidence evaluations are discussed.</p>","PeriodicalId":94012,"journal":{"name":"Forensic science international. Genetics","volume":"80 ","pages":"103336"},"PeriodicalIF":3.1000,"publicationDate":"2026-01-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12382342/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Forensic science international. Genetics","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1016/j.fsigen.2025.103336","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2025/8/5 0:00:00","PubModel":"Epub","JCR":"","JCRName":"","Score":null,"Total":0}
引用次数: 0
Abstract
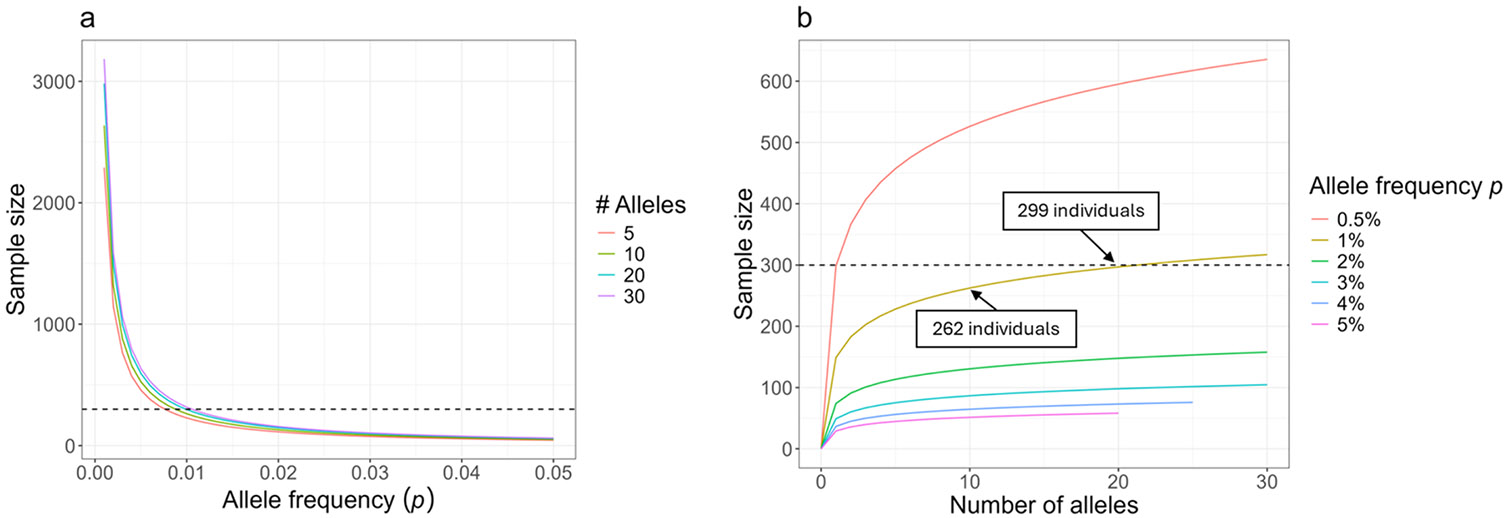
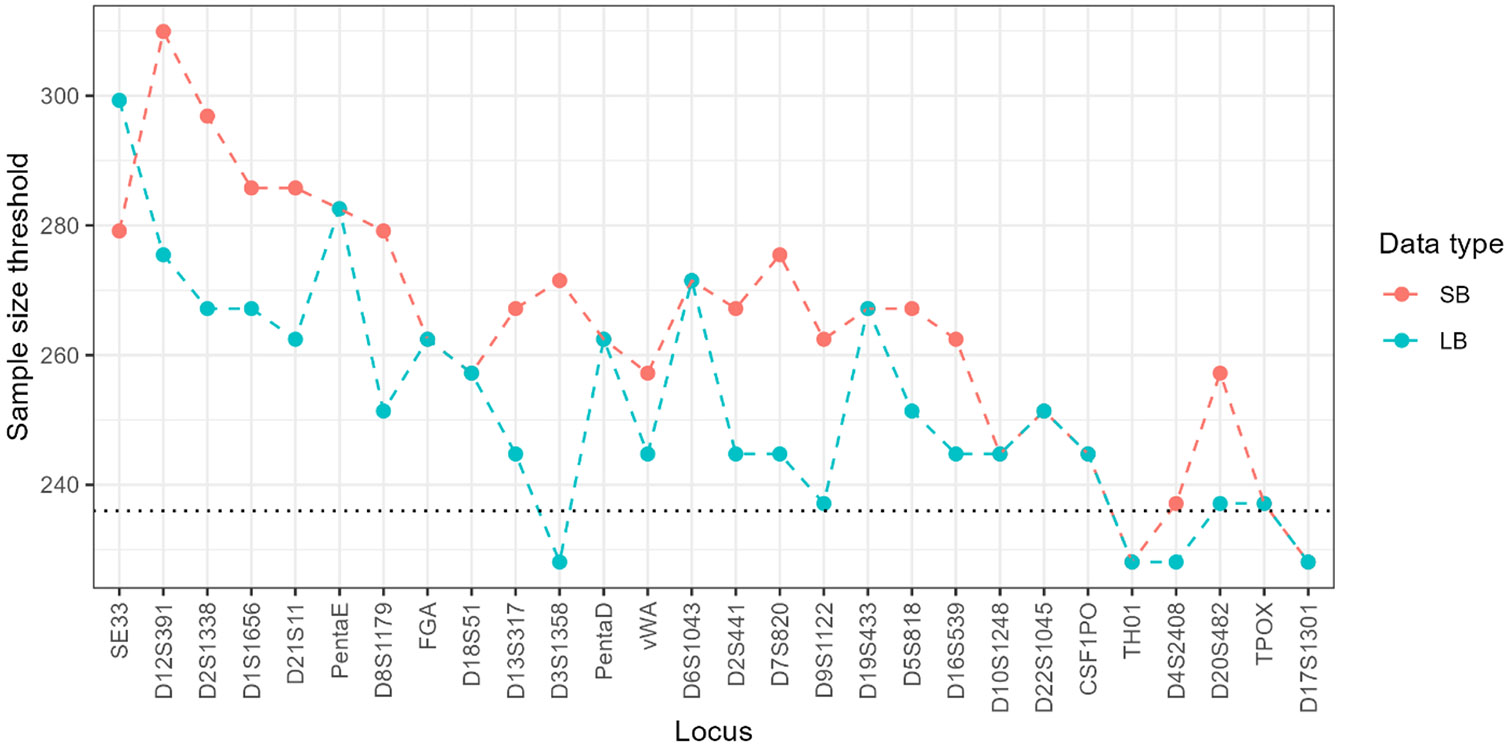
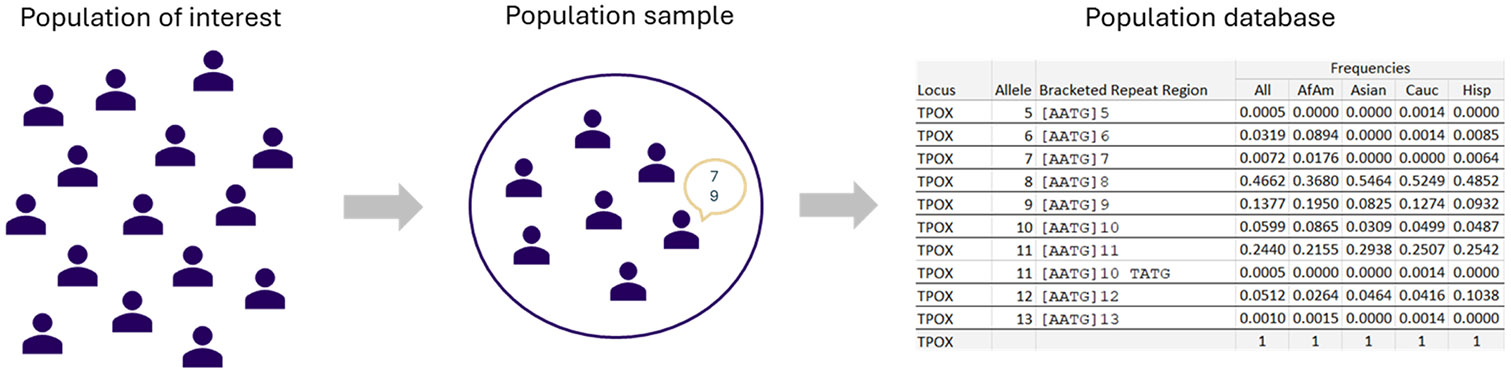
Population databases allow us to attach probabilities to DNA evidence by the estimation of genotype frequencies, which rely on accurate allele frequency estimates. As short tandem repeat (STR) marker sets for human identification have expanded to include more discriminating markers, and especially now that sequencing techniques allow us to distinguish between alleles based on variation in underlying base-pair structure, it is important to reevaluate existing guidance on population database sizes for the estimation of allele frequencies. In this paper, we revisit the topic of population sampling by focusing on the representation of alleles, i.e. whether alleles are observed or not, in a sample of individuals containing data for highly polymorphic autosomal STR loci. The effect of both length- and sequence-based STR data on population sample size implications are demonstrated, and differences between lesser and more polymorphic markers are discussed. The consequences of using a limited number of individuals are explored and the impact of increasing population sample sizes by combining different data sets is shown to help determine the point at which further sampling may no longer provide significant value. Finally, different approaches for accommodating previously unobserved alleles and their impact on DNA evidence evaluations are discussed.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


