Xinhui Li, Shihua Li, Xiao-Jiang Li, Huu Phuc Nguyen, Asa Petersen, Mahmoud A Pouladi
{"title":"The role of oligodendroglial dysfunction in Huntington's disease.","authors":"Xinhui Li, Shihua Li, Xiao-Jiang Li, Huu Phuc Nguyen, Asa Petersen, Mahmoud A Pouladi","doi":"10.1177/18796397251358017","DOIUrl":null,"url":null,"abstract":"<p><p>Huntington's disease (HD) is a fatal neurodegenerative disorder characterized by progressive motor, cognitive, and psychiatric symptoms. Research efforts to understand and treat the disease have historically focused on neuronal pathology, but growing evidence underscores the critical role of oligodendrocytes in its pathogenesis. This review synthesizes recent findings on oligodendroglial dysfunction in HD, showing that white matter abnormalities arise early in disease progression, often preceding gray matter changes and clinical symptoms. Neuroimaging and postmortem studies reveal significant white matter atrophy, myelin breakdown, and impaired oligodendrocyte maturation in both patients and animal models. The myelination response to environmental factors is also altered in HD, suggesting impaired white matter plasticity in the disease. At the molecular level, mutant huntingtin disrupts oligodendrocyte function through transcriptional dysregulation of myelin genes, epigenetic modifications involving PRC2 and REST, altered lipid metabolism, thiamine pathway dysfunction, and aberrant BDNF signaling. Key oligodendroglial transcriptional regulators such as MYRF and TCF7L2 are compromised in HD, leading to defective myelination and reduced metabolic support for neurons. Recognizing the role of these mechanisms provides potential biomarkers for early detection and therapeutic targets aimed at preserving both neuronal and glial function in HD.</p>","PeriodicalId":16042,"journal":{"name":"Journal of Huntington's disease","volume":"14 3","pages":"270-278"},"PeriodicalIF":3.1000,"publicationDate":"2025-08-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12361682/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Huntington's disease","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1177/18796397251358017","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2025/8/7 0:00:00","PubModel":"Epub","JCR":"Q3","JCRName":"NEUROSCIENCES","Score":null,"Total":0}
引用次数: 0
Abstract
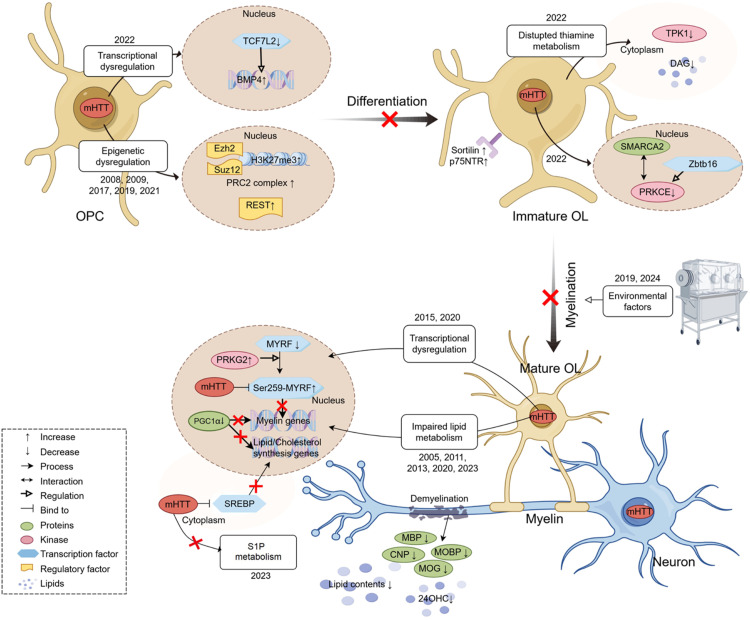
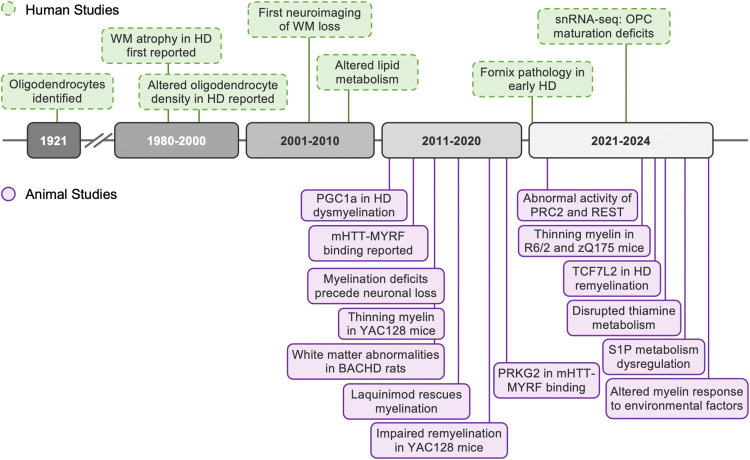
Huntington's disease (HD) is a fatal neurodegenerative disorder characterized by progressive motor, cognitive, and psychiatric symptoms. Research efforts to understand and treat the disease have historically focused on neuronal pathology, but growing evidence underscores the critical role of oligodendrocytes in its pathogenesis. This review synthesizes recent findings on oligodendroglial dysfunction in HD, showing that white matter abnormalities arise early in disease progression, often preceding gray matter changes and clinical symptoms. Neuroimaging and postmortem studies reveal significant white matter atrophy, myelin breakdown, and impaired oligodendrocyte maturation in both patients and animal models. The myelination response to environmental factors is also altered in HD, suggesting impaired white matter plasticity in the disease. At the molecular level, mutant huntingtin disrupts oligodendrocyte function through transcriptional dysregulation of myelin genes, epigenetic modifications involving PRC2 and REST, altered lipid metabolism, thiamine pathway dysfunction, and aberrant BDNF signaling. Key oligodendroglial transcriptional regulators such as MYRF and TCF7L2 are compromised in HD, leading to defective myelination and reduced metabolic support for neurons. Recognizing the role of these mechanisms provides potential biomarkers for early detection and therapeutic targets aimed at preserving both neuronal and glial function in HD.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


