{"title":"GenomicLayers: sequence-based simulation of epi-genomes.","authors":"Dave T Gerrard","doi":"10.1186/s12859-025-06224-y","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>Cellular development and differentiation in Eukaryotes depends upon sequential gene regulatory decisions that allow a single genome to encode many hundreds of distinct cellular phenotypes. Decisions are stored in the regulatory state of each cell, an important part of which is the epi-genome-the collection of proteins, RNA and their specific associations with the genome. Additionally, further cellular responses are, in part, determined by this regulatory state. To date, models of regulatory state have failed to include the contingency of incoming regulatory signals on the current epi-genetic state and none have done so at the whole-genome level.</p><p><strong>Results: </strong>Here we introduce GenomicLayers, a new R package to run rules-based simulations of epigenetic state changes genome-wide in Eukaryotes. Simulations model the accumulation of changes to genome-wide layers by user-specified binding factors. As a first exemplar, we show two versions of a simple model of the recruitment and spreading of epigenetic marks near telomeres in the yeast Saccharomyces cerevisiae. By combining the output from 100 runs of the simulation, we generate whole genome predictions of epigenetic state at 1 bp resolution. The example yeast models are included within a 'vignette' with the GenomicLayers package, which is available at https://github.com/davetgerrard/GenomicLayers . To demonstrate the use of GenomicLayers on the full human reference genome (hg38), we show the results from parameter refinement on a simplistic model of the action of pluripotency factors against a self-spreading repressor seeded at CpG islands. The human genome model is included in supplementary information as an R script.</p><p><strong>Conclusions: </strong>GenomicLayers enables scientists working on diverse eukaryotic organisms to test models of gene regulation in silico. Applications include epigenetic silencing, activation by combinatorial binding of transcription factors and the sink effects caused by down-regulation of components of epigenetic regulators. The software is intended to be used to parameterise, refine and combine models and thereby capitalise on data from the thousands of studies of Eukaryotic epigenomes.</p>","PeriodicalId":8958,"journal":{"name":"BMC Bioinformatics","volume":"26 1","pages":"205"},"PeriodicalIF":3.3000,"publicationDate":"2025-08-04","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12323044/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"BMC Bioinformatics","FirstCategoryId":"99","ListUrlMain":"https://doi.org/10.1186/s12859-025-06224-y","RegionNum":3,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"BIOCHEMICAL RESEARCH METHODS","Score":null,"Total":0}
引用次数: 0
Abstract
Background: Cellular development and differentiation in Eukaryotes depends upon sequential gene regulatory decisions that allow a single genome to encode many hundreds of distinct cellular phenotypes. Decisions are stored in the regulatory state of each cell, an important part of which is the epi-genome-the collection of proteins, RNA and their specific associations with the genome. Additionally, further cellular responses are, in part, determined by this regulatory state. To date, models of regulatory state have failed to include the contingency of incoming regulatory signals on the current epi-genetic state and none have done so at the whole-genome level.
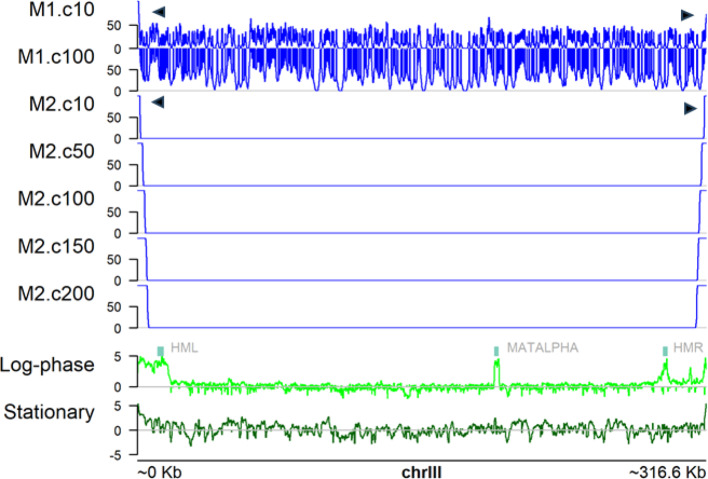
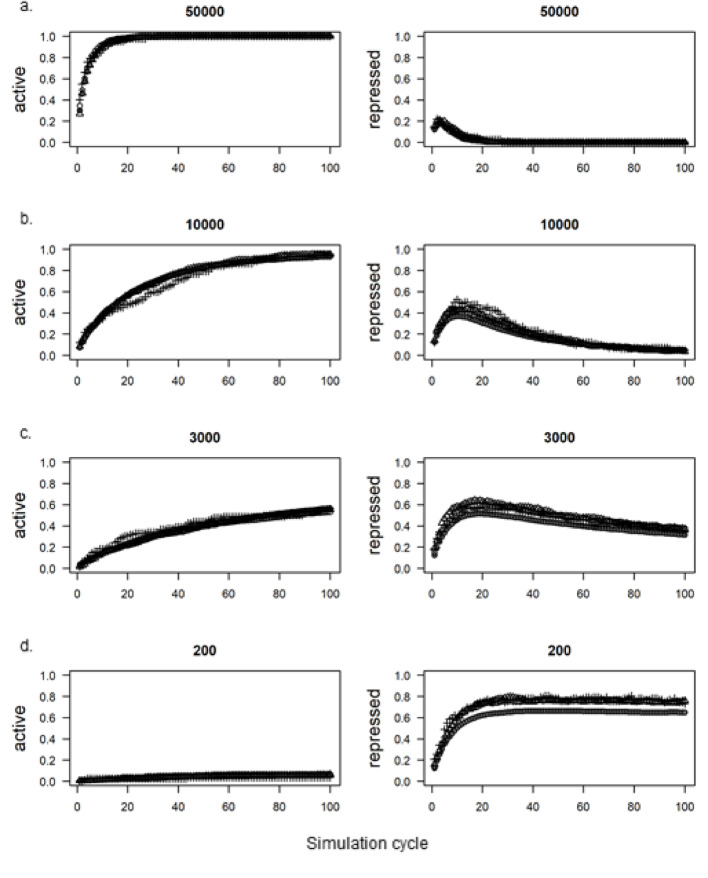
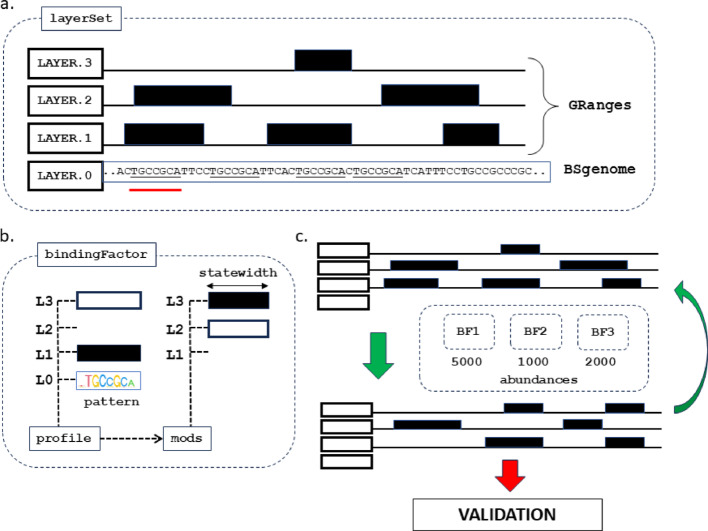
Results: Here we introduce GenomicLayers, a new R package to run rules-based simulations of epigenetic state changes genome-wide in Eukaryotes. Simulations model the accumulation of changes to genome-wide layers by user-specified binding factors. As a first exemplar, we show two versions of a simple model of the recruitment and spreading of epigenetic marks near telomeres in the yeast Saccharomyces cerevisiae. By combining the output from 100 runs of the simulation, we generate whole genome predictions of epigenetic state at 1 bp resolution. The example yeast models are included within a 'vignette' with the GenomicLayers package, which is available at https://github.com/davetgerrard/GenomicLayers . To demonstrate the use of GenomicLayers on the full human reference genome (hg38), we show the results from parameter refinement on a simplistic model of the action of pluripotency factors against a self-spreading repressor seeded at CpG islands. The human genome model is included in supplementary information as an R script.
Conclusions: GenomicLayers enables scientists working on diverse eukaryotic organisms to test models of gene regulation in silico. Applications include epigenetic silencing, activation by combinatorial binding of transcription factors and the sink effects caused by down-regulation of components of epigenetic regulators. The software is intended to be used to parameterise, refine and combine models and thereby capitalise on data from the thousands of studies of Eukaryotic epigenomes.
期刊介绍:
BMC Bioinformatics is an open access, peer-reviewed journal that considers articles on all aspects of the development, testing and novel application of computational and statistical methods for the modeling and analysis of all kinds of biological data, as well as other areas of computational biology.
BMC Bioinformatics is part of the BMC series which publishes subject-specific journals focused on the needs of individual research communities across all areas of biology and medicine. We offer an efficient, fair and friendly peer review service, and are committed to publishing all sound science, provided that there is some advance in knowledge presented by the work.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


