{"title":"49, XXXYY: Parental Origin, Occurrence, and Clinical Phenotypes.","authors":"Yufang Du, Liangrong Liao, Xianda Wei, Yunting Ma, Meizhen Shi, Chunyan Li, Juliang Liu, Wenting Lin, Hao Zeng, Shaoke Chen, Baoheng Gui","doi":"10.1155/genr/1368153","DOIUrl":null,"url":null,"abstract":"<p><p>49, XXXYY is a rare form of sex chromosomal aneuploidy that has been reported in 11 cases worldwide. The parental origin of the extra sex chromosomes and the specific clinical features of this condition remain unclear. We recruited a case with 49, XXXYY and performed genome-wide copy number variation analysis using next-generation sequencing. In addition, the parental origin of the extra sex chromosomes was determined through short tandem repeats (STRs) locus genotyping. Furthermore, a comprehensive review and comparison of clinical phenotypes were conducted among 12 cases with 49, XXXYY. The patient exhibited a karyotype of 49, XXXYY without any mosaic patterns. No pathogenic microdeletions or microduplications (> 100 kb) were identified in autosomes 1-22. Analysis of the STR loci revealed that two of three X chromosomes originated from father. This suggests that the nondisjunction of chromosomes X and Y during stages I and II of meiotic spermatogenesis led to the production of an abnormal sperm with XXYY. Subsequently, fertilization of a normal oocyte with this abnormal sperm resulted in an abnormal zygote with pentasomy XXXYY. The main clinical features observed in these cases included varying degrees of mental retardation, minor facial dysmorphology, and gonadal or endocrine abnormalities. In conclusion, 49, XXXYY is a rare chromosomal disorder characterized by mental retardation and facial dysmorphology. Nondisjunction of chromosomes X and Y during stages I and II of meiotic spermatogenesis is a critical factor contributing to the development of this abnormal karyotype.</p>","PeriodicalId":12778,"journal":{"name":"Genetics research","volume":"2025 ","pages":"1368153"},"PeriodicalIF":2.1000,"publicationDate":"2025-07-21","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12316498/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Genetics research","FirstCategoryId":"99","ListUrlMain":"https://doi.org/10.1155/genr/1368153","RegionNum":4,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2025/1/1 0:00:00","PubModel":"eCollection","JCR":"Q4","JCRName":"GENETICS & HEREDITY","Score":null,"Total":0}
引用次数: 0
Abstract
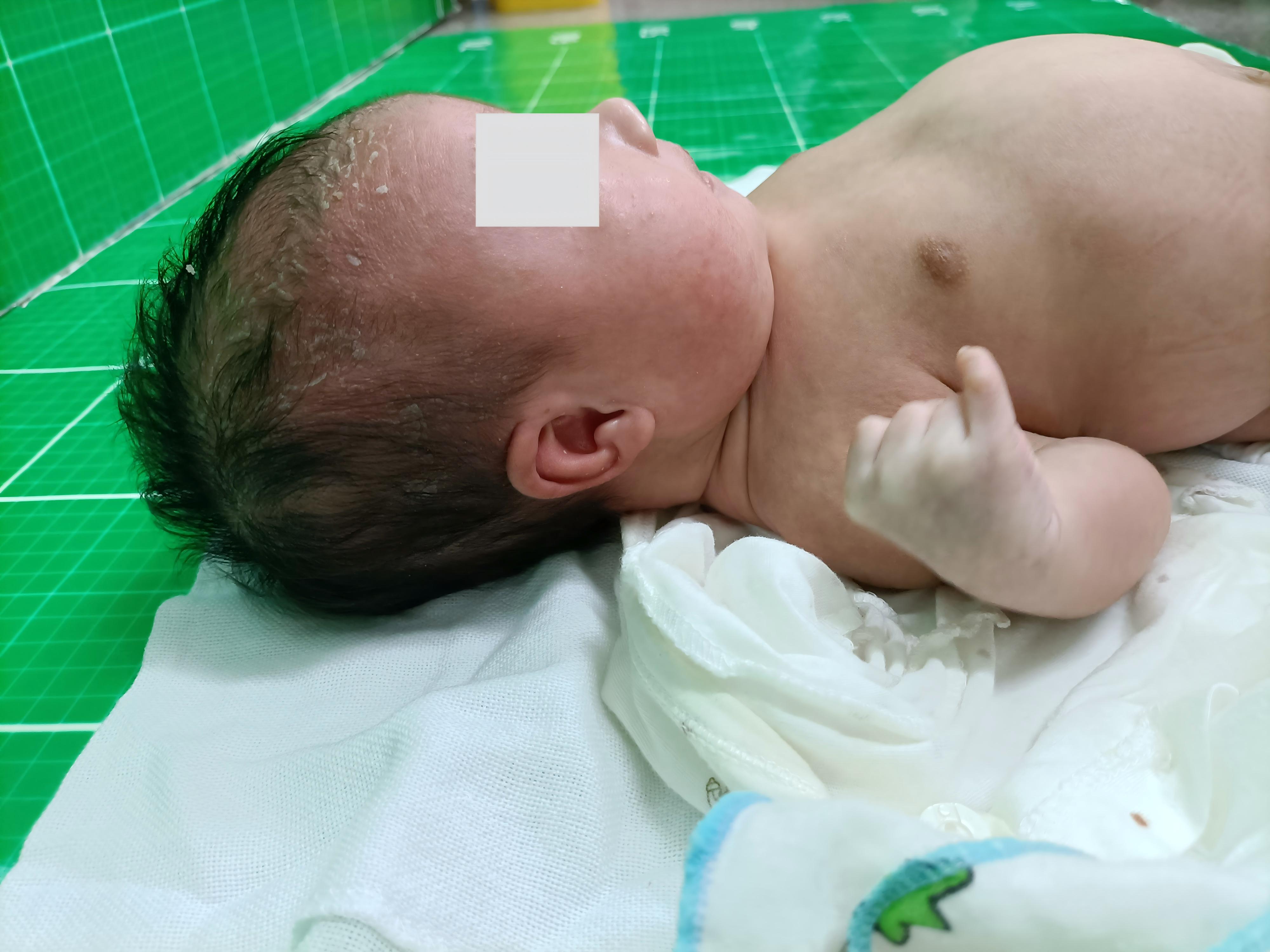
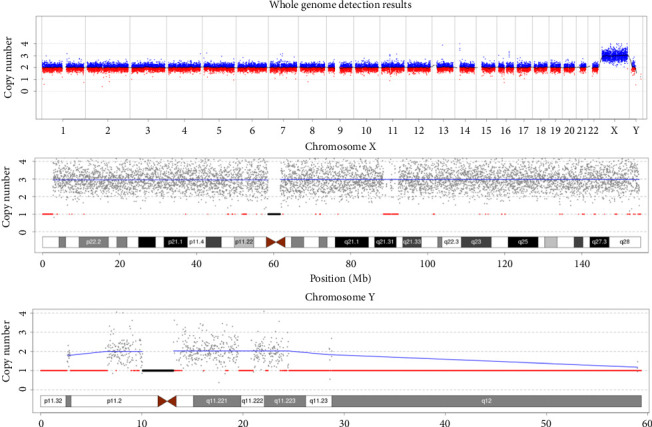
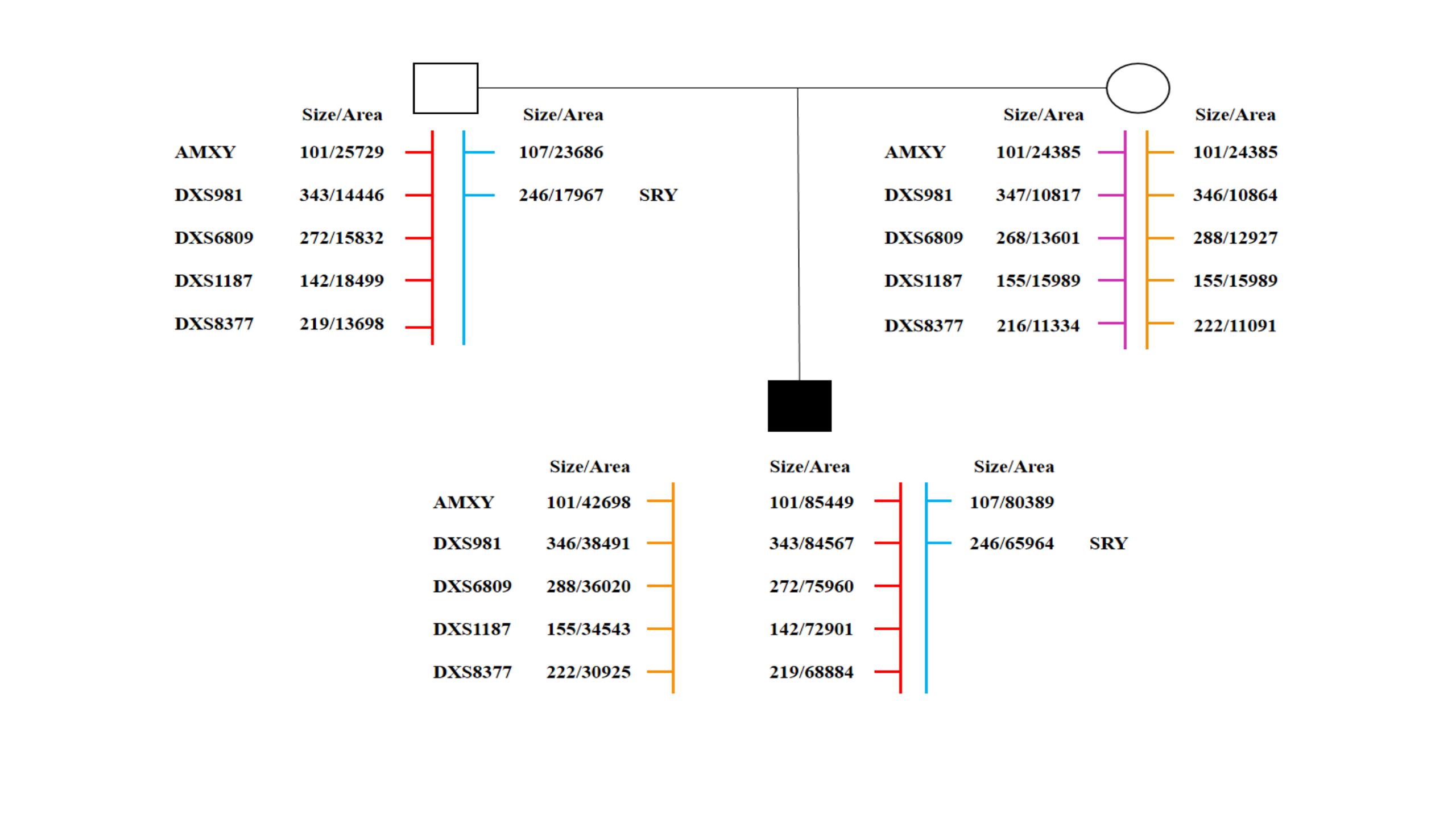
49, XXXYY is a rare form of sex chromosomal aneuploidy that has been reported in 11 cases worldwide. The parental origin of the extra sex chromosomes and the specific clinical features of this condition remain unclear. We recruited a case with 49, XXXYY and performed genome-wide copy number variation analysis using next-generation sequencing. In addition, the parental origin of the extra sex chromosomes was determined through short tandem repeats (STRs) locus genotyping. Furthermore, a comprehensive review and comparison of clinical phenotypes were conducted among 12 cases with 49, XXXYY. The patient exhibited a karyotype of 49, XXXYY without any mosaic patterns. No pathogenic microdeletions or microduplications (> 100 kb) were identified in autosomes 1-22. Analysis of the STR loci revealed that two of three X chromosomes originated from father. This suggests that the nondisjunction of chromosomes X and Y during stages I and II of meiotic spermatogenesis led to the production of an abnormal sperm with XXYY. Subsequently, fertilization of a normal oocyte with this abnormal sperm resulted in an abnormal zygote with pentasomy XXXYY. The main clinical features observed in these cases included varying degrees of mental retardation, minor facial dysmorphology, and gonadal or endocrine abnormalities. In conclusion, 49, XXXYY is a rare chromosomal disorder characterized by mental retardation and facial dysmorphology. Nondisjunction of chromosomes X and Y during stages I and II of meiotic spermatogenesis is a critical factor contributing to the development of this abnormal karyotype.
期刊介绍:
Genetics Research is a key forum for original research on all aspects of human and animal genetics, reporting key findings on genomes, genes, mutations and molecular interactions, extending out to developmental, evolutionary, and population genetics as well as ethical, legal and social aspects. Our aim is to lead to a better understanding of genetic processes in health and disease. The journal focuses on the use of new technologies, such as next generation sequencing together with bioinformatics analysis, to produce increasingly detailed views of how genes function in tissues and how these genes perform, individually or collectively, in normal development and disease aetiology. The journal publishes original work, review articles, short papers, computational studies, and novel methods and techniques in research covering humans and well-established genetic organisms. Key subject areas include medical genetics, genomics, human evolutionary and population genetics, bioinformatics, genetics of complex traits, molecular and developmental genetics, Evo-Devo, quantitative and statistical genetics, behavioural genetics and environmental genetics. The breadth and quality of research make the journal an invaluable resource for medical geneticists, molecular biologists, bioinformaticians and researchers involved in genetic basis of diseases, evolutionary and developmental studies.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


