Protein–peptide docking with a rational and accurate diffusion generative model
IF 23.9
1区 计算机科学
Q1 COMPUTER SCIENCE, ARTIFICIAL INTELLIGENCE
引用次数: 0
Abstract
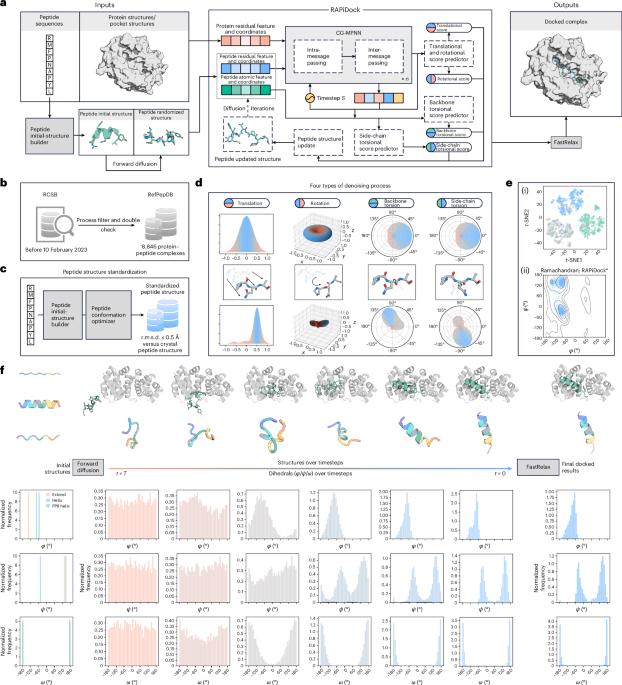
Therapeutic peptides represent the forefront of drug discovery, offering potent and safe alternatives to traditional small molecules. However, their weak and context-dependent nature complicates the efficient virtual screening and structural characterization of protein–peptide patterns. Here we introduce RAPiDock, a diffusion generative model designed for rational, accurate and rapid protein–peptide docking at an all-atomic level. RAPiDock efficiently reduces the sampling space by incorporating physical constraints and uses a bi-scale graph to effectively capture multidimensional structural information while balancing efficiency. In addition, the model uses a Clebsch–Gordan tensor product-based architecture to ensure physical symmetry. RAPiDock outperforms existing tools in prediction of protein–peptide-binding patterns, achieving a 93.7% success rate at top-25 predictions (13.4% higher than AlphaFold2-Multimer), with an execution speed of 0.35 seconds per complex (~270 times faster than AlphaFold2-Multimer). Extensive experiments demonstrate RAPiDock’s remarkable ability to handle 92 types of residue including posttranslational modifications, accurately predict subtle docking patterns, successfully identify multiple potential peptide-binding sites in global docking and serve as a powerful tool for high-throughput virtual screening with structural precision. All these push the boundaries of efficient protein–peptide docking in multiple real-application scenarios. Zhao et al. present RAPiDock, an all-atom diffusion model that predicts peptide–protein binding patterns across 92 amino acid types, enabling high-throughput virtual screening for advancing therapeutic peptide design.

合理准确的扩散生成模型实现蛋白肽对接
治疗性多肽代表了药物发现的前沿,为传统的小分子提供了有效和安全的替代品。然而,它们的弱和上下文依赖性使有效的虚拟筛选和蛋白质-肽模式的结构表征复杂化。RAPiDock是一种在全原子水平上为合理、准确、快速的蛋白肽对接而设计的扩散生成模型。RAPiDock通过结合物理约束有效地减少了采样空间,并使用双尺度图在平衡效率的同时有效地捕获多维结构信息。此外,该模型使用基于Clebsch-Gordan张量积的架构来确保物理对称性。RAPiDock在预测蛋白质-肽结合模式方面优于现有工具,在前25位预测中达到93.7%的成功率(比AlphaFold2-Multimer高13.4%),每个复合物的执行速度为0.35秒(比AlphaFold2-Multimer快270倍)。大量的实验表明,RAPiDock能够处理92种类型的残基,包括翻译后修饰,准确预测微妙的对接模式,成功识别全局对接中的多个潜在肽结合位点,并作为具有结构精度的高通量虚拟筛选的强大工具。这些都在多个实际应用场景中推动了高效蛋白肽对接的边界。
本文章由计算机程序翻译,如有差异,请以英文原文为准。
求助全文
约1分钟内获得全文
求助全文
来源期刊

Nature Machine Intelligence
Multiple-
CiteScore
36.90
自引率
2.10%
发文量
127
期刊介绍:
Nature Machine Intelligence is a distinguished publication that presents original research and reviews on various topics in machine learning, robotics, and AI. Our focus extends beyond these fields, exploring their profound impact on other scientific disciplines, as well as societal and industrial aspects. We recognize limitless possibilities wherein machine intelligence can augment human capabilities and knowledge in domains like scientific exploration, healthcare, medical diagnostics, and the creation of safe and sustainable cities, transportation, and agriculture. Simultaneously, we acknowledge the emergence of ethical, social, and legal concerns due to the rapid pace of advancements.
To foster interdisciplinary discussions on these far-reaching implications, Nature Machine Intelligence serves as a platform for dialogue facilitated through Comments, News Features, News & Views articles, and Correspondence. Our goal is to encourage a comprehensive examination of these subjects.
Similar to all Nature-branded journals, Nature Machine Intelligence operates under the guidance of a team of skilled editors. We adhere to a fair and rigorous peer-review process, ensuring high standards of copy-editing and production, swift publication, and editorial independence.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:



