Theoretical study of the reactivity of oligo-pyrrole derivatives linked at their ends to the Magnesol, Ferrol, and Zinkol fragments: NBO, DFT, and TD-DFT calculations
Mohammed Zenati, Madani Hedidi, Abdelkader M. Elhorri, Hicham Mahdjoub-araibi, Assia Laib
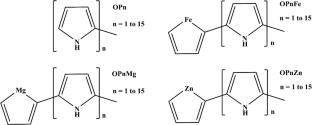
{"title":"Theoretical study of the reactivity of oligo-pyrrole derivatives linked at their ends to the Magnesol, Ferrol, and Zinkol fragments: NBO, DFT, and TD-DFT calculations","authors":"Mohammed Zenati, Madani Hedidi, Abdelkader M. Elhorri, Hicham Mahdjoub-araibi, Assia Laib","doi":"10.1007/s00894-025-06443-3","DOIUrl":null,"url":null,"abstract":"<div><h3>Context</h3><p>This study compares the electronic and structural properties of oligopyrroles (OPn) grafted with metal rings (Magnesol OPnMg, Ferrol OPnFe, and Zinkol OPnZn) with those of standard oligopyrroles (OPn). In the context of the polymerization of oligomers from <i>n</i> = 1 to <i>n</i> = 15, the parameters of global reactivity and excited states obey exponential equations (ƒ(<i>x</i>) = <i>A</i> + <i>B</i> Exp<sup>−<i>R</i>(<i>x</i>)</sup>), allowing values to be predicted to infinity. The corresponding infinity results reveal that energy gaps (Δ<i>E</i><sub>H-L</sub>) vary between 3.14 and 5.20 eV, while chemical hardnesses (<i>η</i>) oscillate between 1.507 and 2.60 eV, with overall electrophilicity (<i>ω</i>) and nucleophilicity (Nu) of 0.74–1.40 eV and 5.47–5.53 eV, respectively. The new oligopyrrole derivatives show increases in intramolecular charge transfer (ICT) and UV–Vis absorptions in the violet (<i>λ</i><sub>max</sub> between 407 and 431 nm). NBO analysis shows a reduction in hydrogen charges in the –NH groups of oligopyrroles based on metal derivatives, enhancing their nucleophilicity. In addition, these molecules display better solubility, with favorable solvation energies (Δ<i>G</i><sub>solv</sub>) (24–35 kcal-mol<sup>−1</sup>) compared to OPn. Finally, metal-based derivatives show stronger interactions with formaldehyde (HCHO) in aqueous media than OPn, demonstrating interaction energy differences (<i>E</i><sub>int</sub>) ranging from 0.7 to 1.40 kcal-mol<sup>−1</sup>.</p><h3>Methods</h3><p>All calculations were performed using the Gaussian 16 program. The selected functionals are: B3LYP, CAM–BLYP, B3LYP–D3, CAM–BLYP–D3, and TD–CAM–BLYP. The basis–sets used are: 6–31 + + G(d,p) and LanL2DZ. Finally, the natural bond orbital (NBO) method was used in this study.</p></div>","PeriodicalId":651,"journal":{"name":"Journal of Molecular Modeling","volume":"31 8","pages":""},"PeriodicalIF":2.5000,"publicationDate":"2025-08-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Molecular Modeling","FirstCategoryId":"92","ListUrlMain":"https://link.springer.com/article/10.1007/s00894-025-06443-3","RegionNum":4,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q4","JCRName":"BIOCHEMISTRY & MOLECULAR BIOLOGY","Score":null,"Total":0}
引用次数: 0
Abstract
Context
This study compares the electronic and structural properties of oligopyrroles (OPn) grafted with metal rings (Magnesol OPnMg, Ferrol OPnFe, and Zinkol OPnZn) with those of standard oligopyrroles (OPn). In the context of the polymerization of oligomers from n = 1 to n = 15, the parameters of global reactivity and excited states obey exponential equations (ƒ(x) = A + B Exp−R(x)), allowing values to be predicted to infinity. The corresponding infinity results reveal that energy gaps (ΔEH-L) vary between 3.14 and 5.20 eV, while chemical hardnesses (η) oscillate between 1.507 and 2.60 eV, with overall electrophilicity (ω) and nucleophilicity (Nu) of 0.74–1.40 eV and 5.47–5.53 eV, respectively. The new oligopyrrole derivatives show increases in intramolecular charge transfer (ICT) and UV–Vis absorptions in the violet (λmax between 407 and 431 nm). NBO analysis shows a reduction in hydrogen charges in the –NH groups of oligopyrroles based on metal derivatives, enhancing their nucleophilicity. In addition, these molecules display better solubility, with favorable solvation energies (ΔGsolv) (24–35 kcal-mol−1) compared to OPn. Finally, metal-based derivatives show stronger interactions with formaldehyde (HCHO) in aqueous media than OPn, demonstrating interaction energy differences (Eint) ranging from 0.7 to 1.40 kcal-mol−1.
Methods
All calculations were performed using the Gaussian 16 program. The selected functionals are: B3LYP, CAM–BLYP, B3LYP–D3, CAM–BLYP–D3, and TD–CAM–BLYP. The basis–sets used are: 6–31 + + G(d,p) and LanL2DZ. Finally, the natural bond orbital (NBO) method was used in this study.
期刊介绍:
The Journal of Molecular Modeling focuses on "hardcore" modeling, publishing high-quality research and reports. Founded in 1995 as a purely electronic journal, it has adapted its format to include a full-color print edition, and adjusted its aims and scope fit the fast-changing field of molecular modeling, with a particular focus on three-dimensional modeling.
Today, the journal covers all aspects of molecular modeling including life science modeling; materials modeling; new methods; and computational chemistry.
Topics include computer-aided molecular design; rational drug design, de novo ligand design, receptor modeling and docking; cheminformatics, data analysis, visualization and mining; computational medicinal chemistry; homology modeling; simulation of peptides, DNA and other biopolymers; quantitative structure-activity relationships (QSAR) and ADME-modeling; modeling of biological reaction mechanisms; and combined experimental and computational studies in which calculations play a major role.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


