{"title":"Estimating allele frequencies, ancestry proportions and genotype likelihoods in the presence of mapping bias.","authors":"Torsten Günther, Amy Goldberg, Joshua G Schraiber","doi":"10.1093/g3journal/jkaf172","DOIUrl":null,"url":null,"abstract":"<p><p>Population genomic analyses rely on an accurate and unbiased characterization of the genetic composition of the studied population. For short-read, high-throughput sequencing data, mapping sequencing reads to a linear reference genome can bias population genetic inference due to mismatches in reads carrying non-reference alleles. In this study, we investigate the impact of mapping bias on allele frequency estimates from pseudohaploid data and genotype likelihoods, 2 approaches commonly used in ultra-low to medium coverage sequencing. To mitigate mapping bias, we propose an empirical adjustment to genotype likelihoods. Using data from the 1000 Genomes Project, we find that our new method improves allele frequency estimation. To test a downstream application, we simulate ancient DNA data with realistic post-mortem damage to compare widely used methods for estimating ancestry proportions under different scenarios, including reference genome selection, population divergence, and sequencing depth. Our findings reveal that mapping bias can lead to differences in estimated admixture proportion of up to 4% depending on the reference population. However, the choice of method has a much stronger impact, with some methods showing differences of 10%. qpAdm appears to perform best at estimating simulated ancestry proportions, but it is sensitive to mapping bias and its applicability may vary across species due to its requirement for additional populations beyond the sources and target population. Our adjusted genotype likelihood approach largely mitigates the effect of mapping bias on genome-wide ancestry estimates from genotype likelihood-based tools. However, it cannot account for the bias introduced by the method itself or the noise in individual site allele frequency estimates due to low sequencing depth. Overall, our study provides valuable insights for obtaining more precise estimates of allele frequencies and ancestry proportions in empirical studies.</p>","PeriodicalId":12468,"journal":{"name":"G3: Genes|Genomes|Genetics","volume":" ","pages":""},"PeriodicalIF":2.2000,"publicationDate":"2025-10-08","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12506655/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"G3: Genes|Genomes|Genetics","FirstCategoryId":"99","ListUrlMain":"https://doi.org/10.1093/g3journal/jkaf172","RegionNum":3,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"GENETICS & HEREDITY","Score":null,"Total":0}
引用次数: 0
Abstract
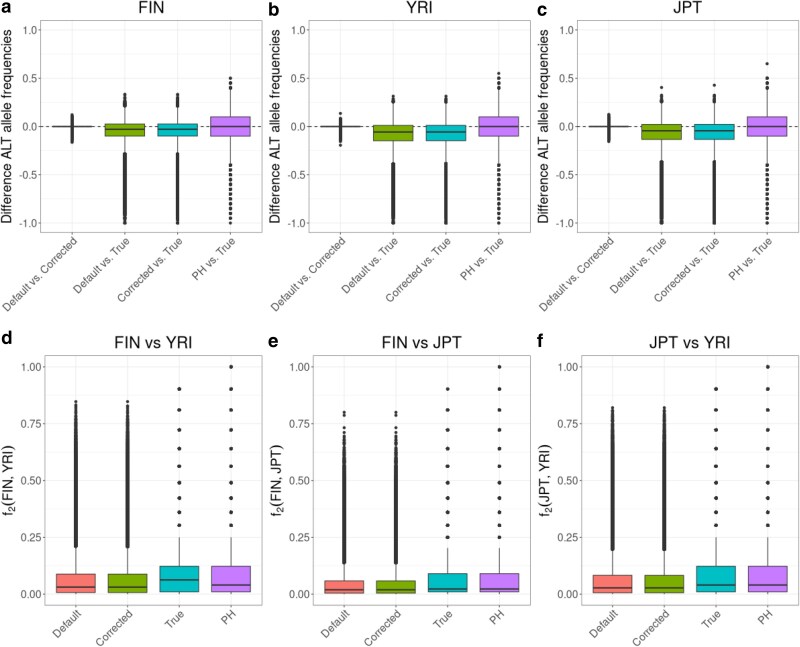
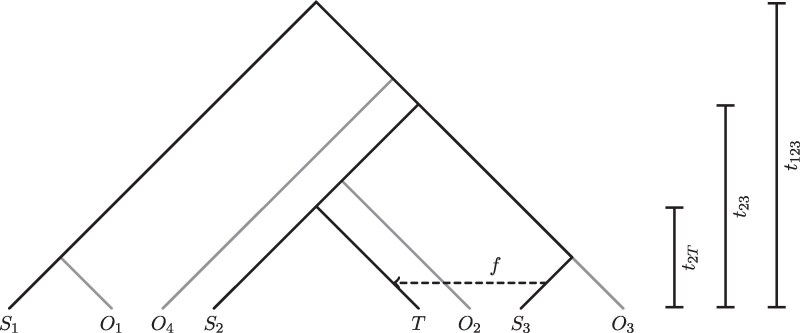
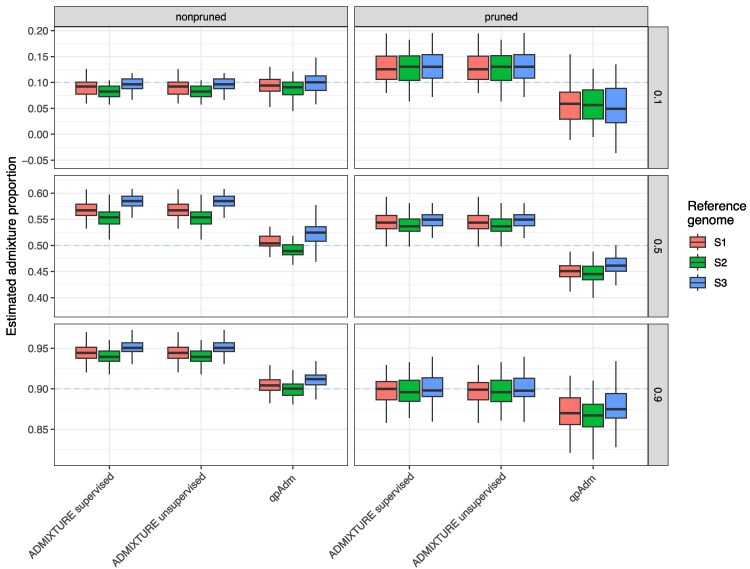
Population genomic analyses rely on an accurate and unbiased characterization of the genetic composition of the studied population. For short-read, high-throughput sequencing data, mapping sequencing reads to a linear reference genome can bias population genetic inference due to mismatches in reads carrying non-reference alleles. In this study, we investigate the impact of mapping bias on allele frequency estimates from pseudohaploid data and genotype likelihoods, 2 approaches commonly used in ultra-low to medium coverage sequencing. To mitigate mapping bias, we propose an empirical adjustment to genotype likelihoods. Using data from the 1000 Genomes Project, we find that our new method improves allele frequency estimation. To test a downstream application, we simulate ancient DNA data with realistic post-mortem damage to compare widely used methods for estimating ancestry proportions under different scenarios, including reference genome selection, population divergence, and sequencing depth. Our findings reveal that mapping bias can lead to differences in estimated admixture proportion of up to 4% depending on the reference population. However, the choice of method has a much stronger impact, with some methods showing differences of 10%. qpAdm appears to perform best at estimating simulated ancestry proportions, but it is sensitive to mapping bias and its applicability may vary across species due to its requirement for additional populations beyond the sources and target population. Our adjusted genotype likelihood approach largely mitigates the effect of mapping bias on genome-wide ancestry estimates from genotype likelihood-based tools. However, it cannot account for the bias introduced by the method itself or the noise in individual site allele frequency estimates due to low sequencing depth. Overall, our study provides valuable insights for obtaining more precise estimates of allele frequencies and ancestry proportions in empirical studies.
期刊介绍:
G3: Genes, Genomes, Genetics provides a forum for the publication of high‐quality foundational research, particularly research that generates useful genetic and genomic information such as genome maps, single gene studies, genome‐wide association and QTL studies, as well as genome reports, mutant screens, and advances in methods and technology. The Editorial Board of G3 believes that rapid dissemination of these data is the necessary foundation for analysis that leads to mechanistic insights.
G3, published by the Genetics Society of America, meets the critical and growing need of the genetics community for rapid review and publication of important results in all areas of genetics. G3 offers the opportunity to publish the puzzling finding or to present unpublished results that may not have been submitted for review and publication due to a perceived lack of a potential high-impact finding. G3 has earned the DOAJ Seal, which is a mark of certification for open access journals, awarded by DOAJ to journals that achieve a high level of openness, adhere to Best Practice and high publishing standards.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


