Deciphering Long-Range Effects of Mutations: An Integrated Approach Using Elastic Network Models and Protein Structure Networks
IF 4.5
2区 生物学
Q1 BIOCHEMISTRY & MOLECULAR BIOLOGY
引用次数: 0
Abstract
Understanding the impact of genetic variants on protein structure and function is essential for deciphering disease mechanisms. The MAVISp framework offers a systematic approach for evaluating structural effects, including variants with long-range impact. In this study, we critically evaluate and refine the LONG_RANGE module of MAVISp, leveraging data from over 400 proteins to optimize parameters for detecting significant response sites. We implement a systematic filtering workflow integrating allosteric free energy, distance constraints, solvent accessibility, and pocket localization to prioritize biologically relevant variants. We benchmarked the results against experimental data from deep mutational scans to identify the optimal combination of thresholds and filtering steps for assessing the impact of allosteric variants at response sites. Our analysis reveals that a 5.5 Å distance threshold, based on atomic distances, effectively minimizes the occurrence of local contacts in the allosteric map while preserving long-range effects. To address the limitations of the elastic network model for predicting allosteric free energy changes in non-globular proteins, we propose introducing three different metrics to assess protein globularity within the MAVISp framework, thereby supporting the design of the trimming to be applied to the input structure. Furthermore, we illustrate the potential of incorporating molecular dynamics simulations and algorithms for path analysis to confirm pairs of allosteric mutation sites and response sites involved in distal communication. Overall, we established a robust and scalable workflow for detecting allosteric protein variants, offering insights into structural communication and disease-associated protein variants.
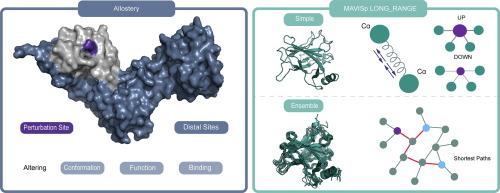
破译突变的长期影响:使用弹性网络模型和蛋白质结构网络的综合方法。
了解遗传变异对蛋白质结构和功能的影响对于破译疾病机制至关重要。MAVISp框架提供了评估结构影响的系统方法,包括具有长期影响的变体。在这项研究中,我们批判性地评估和完善MAVISp的LONG_RANGE模块,利用超过400种蛋白质的数据来优化参数,以检测重要的反应位点。我们实现了一个系统的过滤工作流程,集成了变构自由能、距离限制、溶剂可及性和口袋定位,以优先考虑生物相关的变异。我们将结果与来自深度突变扫描的实验数据进行基准比较,以确定评估响应位点变构变异影响的阈值和过滤步骤的最佳组合。我们的分析表明,基于原子距离的5.5 Å距离阈值有效地减少了变构图中局部接触的发生,同时保留了远程影响。为了解决弹性网络模型在预测非球形蛋白质的变构自由能变化方面的局限性,我们建议在MAVISp框架内引入三种不同的指标来评估蛋白质的球形性,从而支持将修剪应用于输入结构的设计。此外,我们还说明了结合分子动力学模拟和路径分析算法来确认远端通信中涉及的变构突变位点和响应位点对的潜力。总的来说,我们建立了一个强大的、可扩展的工作流程来检测变构蛋白变异,为结构通讯和疾病相关蛋白变异提供了见解。
本文章由计算机程序翻译,如有差异,请以英文原文为准。
求助全文
约1分钟内获得全文
求助全文
来源期刊

Journal of Molecular Biology
生物-生化与分子生物学
CiteScore
11.30
自引率
1.80%
发文量
412
审稿时长
28 days
期刊介绍:
Journal of Molecular Biology (JMB) provides high quality, comprehensive and broad coverage in all areas of molecular biology. The journal publishes original scientific research papers that provide mechanistic and functional insights and report a significant advance to the field. The journal encourages the submission of multidisciplinary studies that use complementary experimental and computational approaches to address challenging biological questions.
Research areas include but are not limited to: Biomolecular interactions, signaling networks, systems biology; Cell cycle, cell growth, cell differentiation; Cell death, autophagy; Cell signaling and regulation; Chemical biology; Computational biology, in combination with experimental studies; DNA replication, repair, and recombination; Development, regenerative biology, mechanistic and functional studies of stem cells; Epigenetics, chromatin structure and function; Gene expression; Membrane processes, cell surface proteins and cell-cell interactions; Methodological advances, both experimental and theoretical, including databases; Microbiology, virology, and interactions with the host or environment; Microbiota mechanistic and functional studies; Nuclear organization; Post-translational modifications, proteomics; Processing and function of biologically important macromolecules and complexes; Molecular basis of disease; RNA processing, structure and functions of non-coding RNAs, transcription; Sorting, spatiotemporal organization, trafficking; Structural biology; Synthetic biology; Translation, protein folding, chaperones, protein degradation and quality control.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


