Sriram Vijendran, Tavis Anderson, Alexey Markin, Oliver Eulenstein
{"title":"Phylo-rs: an extensible phylogenetic analysis library in rust.","authors":"Sriram Vijendran, Tavis Anderson, Alexey Markin, Oliver Eulenstein","doi":"10.1186/s12859-025-06234-w","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>The advent of next-generation and long-read sequencing technologies has provided an ever-increasing wealth of phylogenetic data that require specially designed algorithms to decipher the underlying evolutionary relationships. As large-scale data become increasingly accessible, there is a concomitant need for efficient computational libraries that facilitate the development and dissemination of specialized algorithms for phylogenetic comparative biology.</p><p><strong>Results: </strong>We introduce Phylo-rs: a fast, extensible, general-purpose library for phylogenetic analysis and inference written in the Rust programming language. Phylo-rs leverages a combination of speed, memory-safety, and native WebAssembly support offered by Rust to provide a robust set of memory-efficient data structures and elementary phylogenetic algorithms. Phylo-rs focuses on the efficient and convenient deployment of software aimed at large-scale phylogenetic analysis and inference. Scalability analysis against popular libraries shows that Phylo-rs performs comparably or better on key algorithms. We utilized it to assess the phylogenetic diversity of influenza A virus in swine, identifying virus groups that are undergoing evolutionary expansion that could be targeted for control through multivalent vaccines. Additionally, we used Phylo-rs to enhance phylogenetic inference by visualizing tree space from Markov chain Monte Carlo (MCMC) Bayesian analysis, efficiently computing approximately five billion tree pair distances to evaluate convergence and select MCMC runs for genomic epidemiology.</p><p><strong>Conclusion: </strong>Phylo-rs enables the design and implementation of cutting-edge software for phylogenetic analysis, thereby facilitating the application and dissemination of theoretical advancements in biology. Phylo-rs is available under an open-source license on GitHub at https://github.com/sriram98v/phylo-rs , with documentation available at https://docs.rs/phylo/latest/phylo/ .</p>","PeriodicalId":8958,"journal":{"name":"BMC Bioinformatics","volume":"26 1","pages":"197"},"PeriodicalIF":3.3000,"publicationDate":"2025-07-29","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12309125/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"BMC Bioinformatics","FirstCategoryId":"99","ListUrlMain":"https://doi.org/10.1186/s12859-025-06234-w","RegionNum":3,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"BIOCHEMICAL RESEARCH METHODS","Score":null,"Total":0}
引用次数: 0
Abstract
Background: The advent of next-generation and long-read sequencing technologies has provided an ever-increasing wealth of phylogenetic data that require specially designed algorithms to decipher the underlying evolutionary relationships. As large-scale data become increasingly accessible, there is a concomitant need for efficient computational libraries that facilitate the development and dissemination of specialized algorithms for phylogenetic comparative biology.
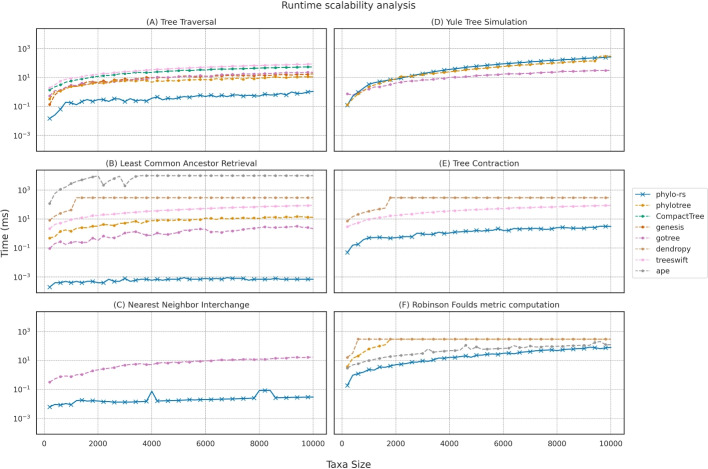
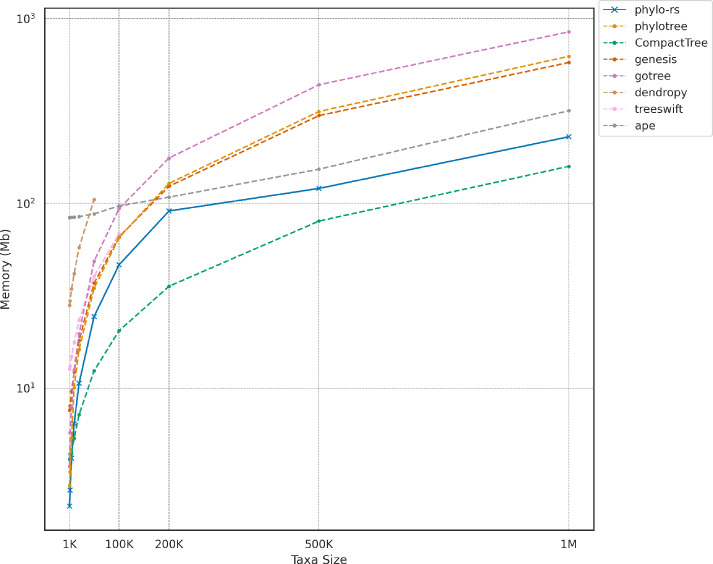
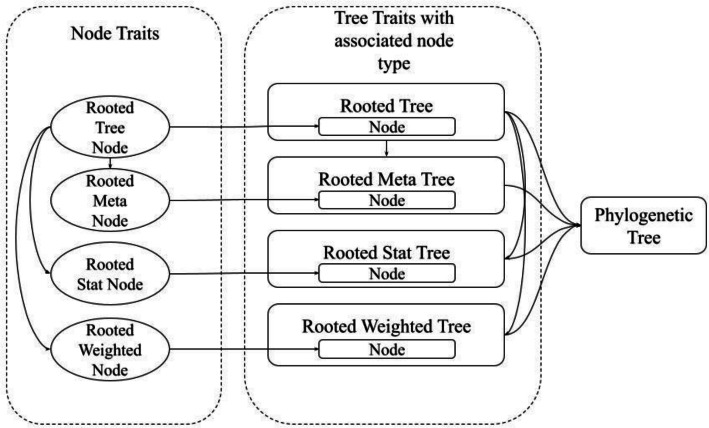
Results: We introduce Phylo-rs: a fast, extensible, general-purpose library for phylogenetic analysis and inference written in the Rust programming language. Phylo-rs leverages a combination of speed, memory-safety, and native WebAssembly support offered by Rust to provide a robust set of memory-efficient data structures and elementary phylogenetic algorithms. Phylo-rs focuses on the efficient and convenient deployment of software aimed at large-scale phylogenetic analysis and inference. Scalability analysis against popular libraries shows that Phylo-rs performs comparably or better on key algorithms. We utilized it to assess the phylogenetic diversity of influenza A virus in swine, identifying virus groups that are undergoing evolutionary expansion that could be targeted for control through multivalent vaccines. Additionally, we used Phylo-rs to enhance phylogenetic inference by visualizing tree space from Markov chain Monte Carlo (MCMC) Bayesian analysis, efficiently computing approximately five billion tree pair distances to evaluate convergence and select MCMC runs for genomic epidemiology.
Conclusion: Phylo-rs enables the design and implementation of cutting-edge software for phylogenetic analysis, thereby facilitating the application and dissemination of theoretical advancements in biology. Phylo-rs is available under an open-source license on GitHub at https://github.com/sriram98v/phylo-rs , with documentation available at https://docs.rs/phylo/latest/phylo/ .
期刊介绍:
BMC Bioinformatics is an open access, peer-reviewed journal that considers articles on all aspects of the development, testing and novel application of computational and statistical methods for the modeling and analysis of all kinds of biological data, as well as other areas of computational biology.
BMC Bioinformatics is part of the BMC series which publishes subject-specific journals focused on the needs of individual research communities across all areas of biology and medicine. We offer an efficient, fair and friendly peer review service, and are committed to publishing all sound science, provided that there is some advance in knowledge presented by the work.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


