Gene editing for Spinocerebellar ataxia type 3 taking advantage of the human ATXN3L paralog as replacement gene
IF 4.5
3区 医学
Q1 BIOCHEMISTRY & MOLECULAR BIOLOGY
引用次数: 0
Abstract
Spinocerebellar ataxia type 3 (SCA3) is a rare neurodegenerative disease caused by a CAG expansion of the ataxin-3 gene (ATXN3). SCA3 patients suffer from ataxia, spasticity and dystonia in mid-adulthood, with spinocerebellar dysfunction and degeneration. As a monogenic disease for which only symptomatic treatment is available, ATXN3 is an attractive target for gene editing. We used the KamiCas9, a self-inactivating gene editing system, to explore gene editing strategies suitable for all SCA3 patients. We first tested the deletion of exon 10 or the introduction of a premature stop codon into exon 9. High editing events were observed in vitro, but efficiency was very low in SCA3 transgenic mice. We then evaluated an ablate-and-replace strategy. The ablate experiments resulted in 55 ± 18% cerebellar editing of the ATXN3 gene. A human ATXN3L paralog, expressed in the brains of SCA3 patients, may act as a natural, CRISPR-resistant replacement gene. In a proof-of-principle study, ablate and ablate-and-replace strategies were evaluated in SCA3 transgenic mice. Two months after injection, similar editing efficiencies were obtained in the ablate and ablate-and-replace groups. Immunofluorescence and RT-qPCR analyses of cerebellar markers support the development of this strategy for SCA3 treatment.
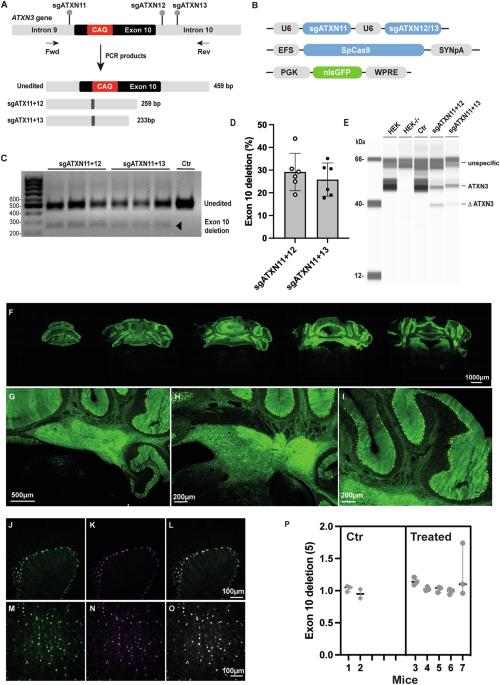
利用人类ATXN3L平行体作为替代基因编辑脊髓小脑性共济失调3型基因。
脊髓小脑性共济失调3型(SCA3)是一种罕见的神经退行性疾病,由ataxin-3基因(ATXN3)的CAG扩增引起。SCA3患者在成年中期出现共济失调、痉挛和肌张力障碍,伴有脊髓小脑功能障碍和变性。作为一种只有对症治疗的单基因疾病,ATXN3是基因编辑的一个有吸引力的靶标。我们使用KamiCas9(一种自我失活的基因编辑系统)来探索适合所有SCA3患者的基因编辑策略。我们首先测试了外显子10的缺失或在外显子9中引入过早终止密码子。在体外观察到高编辑事件,但在SCA3转基因小鼠中效率非常低。然后,我们评估了一个消融和替换策略。消融实验导致55±18%的ATXN3基因在小脑中被编辑。在SCA3患者的大脑中表达的人类ATXN3L类似物可能作为一种天然的抗crispr替代基因。在一项原理验证研究中,在SCA3转基因小鼠中评估了消融和消融-替换策略。注射后2个月,消融组和消融置换组获得了相似的编辑效率。小脑标记物的免疫荧光和RT-qPCR分析支持SCA3治疗策略的发展。
本文章由计算机程序翻译,如有差异,请以英文原文为准。
求助全文
约1分钟内获得全文
求助全文
来源期刊

Gene Therapy
医学-生化与分子生物学
CiteScore
9.70
自引率
2.00%
发文量
67
审稿时长
4-8 weeks
期刊介绍:
Gene Therapy covers both the research and clinical applications of novel therapeutic techniques based on a genetic component. Over the last few decades, significant advances in technologies ranging from identifying novel genetic targets that cause disease through to clinical studies, which show therapeutic benefit, have elevated this multidisciplinary field to the forefront of modern medicine.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


