Mona Nourbakhsh, Mohammad Miryounesi, Ali Tale, Parvaneh Karimzadeh, Hossein Sadeghi, Mohammad-Reza Ghasemi, Nasrin Alipour, Elham Pourbakhtyaran, Nakisa Hooman, Maryam Razzaghy-Azar, Mitra Nourbakhsh, Lil Klaas, Daniel Schulke, Jörn Oliver Sass
{"title":"Glycine N-Acyltransferase Deficiency due to a Homozygous Nonsense Variant in the GLYAT: A Novel Inborn Error of Metabolism","authors":"Mona Nourbakhsh, Mohammad Miryounesi, Ali Tale, Parvaneh Karimzadeh, Hossein Sadeghi, Mohammad-Reza Ghasemi, Nasrin Alipour, Elham Pourbakhtyaran, Nakisa Hooman, Maryam Razzaghy-Azar, Mitra Nourbakhsh, Lil Klaas, Daniel Schulke, Jörn Oliver Sass","doi":"10.1002/jmd2.70032","DOIUrl":null,"url":null,"abstract":"<p>The enzyme glycine <i>N</i>-acyltransferase (GLYAT) plays a crucial role in detoxifying both xenobiotic and endogenous compounds that contain a carboxylic acid group, such as benzoic acid. Data on the impact of human GLYAT on the glycine conjugation pathway is limited and difficult to determine. In this study, we present a 5.7-year-old girl with gross motor delay first noticed at age 5 months and speech delay evident at the time of diagnosis. To the best of our knowledge, no case of GLYAT enzyme deficiency has been reported to date. Whole exome sequencing (WES) identified a homozygous nonsense variant (NM_201648.3: c.322C>T: p.(Q108Ter)) in the <i>GLYAT</i> that abolished GLYAT activity in vitro. The detected variant was confirmed by Sanger sequencing. The patient was treated with pantothenic acid and a mitochondrial cocktail consisting of coenzyme Q10, vitamins B1, B2, B6, B12, C, folate, and carnitine, together with a low-protein diet, which led to the alleviation of edema and hypotonia and an improvement in her motor function and social interactions. Her serum glycine level was also normalized. This case identifies a novel homozygous nonsense variant in the <i>GLYAT</i>, leading to glycine <i>N</i>-acyltransferase enzyme deficiency and associated developmental delays.</p>","PeriodicalId":14930,"journal":{"name":"JIMD reports","volume":"66 5","pages":""},"PeriodicalIF":1.8000,"publicationDate":"2025-07-29","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://onlinelibrary.wiley.com/doi/epdf/10.1002/jmd2.70032","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"JIMD reports","FirstCategoryId":"1085","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1002/jmd2.70032","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"Biochemistry, Genetics and Molecular Biology","Score":null,"Total":0}
引用次数: 0
Abstract
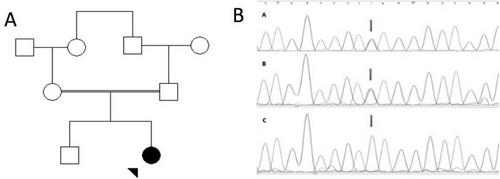
The enzyme glycine N-acyltransferase (GLYAT) plays a crucial role in detoxifying both xenobiotic and endogenous compounds that contain a carboxylic acid group, such as benzoic acid. Data on the impact of human GLYAT on the glycine conjugation pathway is limited and difficult to determine. In this study, we present a 5.7-year-old girl with gross motor delay first noticed at age 5 months and speech delay evident at the time of diagnosis. To the best of our knowledge, no case of GLYAT enzyme deficiency has been reported to date. Whole exome sequencing (WES) identified a homozygous nonsense variant (NM_201648.3: c.322C>T: p.(Q108Ter)) in the GLYAT that abolished GLYAT activity in vitro. The detected variant was confirmed by Sanger sequencing. The patient was treated with pantothenic acid and a mitochondrial cocktail consisting of coenzyme Q10, vitamins B1, B2, B6, B12, C, folate, and carnitine, together with a low-protein diet, which led to the alleviation of edema and hypotonia and an improvement in her motor function and social interactions. Her serum glycine level was also normalized. This case identifies a novel homozygous nonsense variant in the GLYAT, leading to glycine N-acyltransferase enzyme deficiency and associated developmental delays.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


