Andrew Carroll, Alexey Kolesnikov, Daniel E Cook, Lucas Brambrink, Kelly N Wiseman, Sophie M Billings, Semyon Kruglyak, Bryan R Lajoie, Junhua Zhao, Shawn E Levy, Cory Y McLean, Kishwar Shafin, Maria Nattestad, Pi-Chuan Chang
{"title":"Accurate human genome analysis with element avidity sequencing.","authors":"Andrew Carroll, Alexey Kolesnikov, Daniel E Cook, Lucas Brambrink, Kelly N Wiseman, Sophie M Billings, Semyon Kruglyak, Bryan R Lajoie, Junhua Zhao, Shawn E Levy, Cory Y McLean, Kishwar Shafin, Maria Nattestad, Pi-Chuan Chang","doi":"10.1186/s12859-025-06191-4","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>New sequencing technologies provide options for the scientific community to design studies and build clinical workflows. These options expand user choice, and can enable more accurate, scalable, or affordable workflows depending on the fit between scientist needs and platform capability. However, it is essential to understand the performance of these new technologies for different tasks, especially for capabilities that were not possible or tractable in prior technologies. We investigate the new sequencing technology avidity from Element Biosciences. to help the scientific community understand the performance of the options to generate sequencing data.</p><p><strong>Results: </strong>We show that Element whole genome sequencing achieves higher mapping and variant calling accuracy compared to Illumina sequencing at the same coverage, with larger differences at lower coverages (20-30x). We quantify base error rates of Element reads, finding lower error rates, especially in homopolymer and tandem repeat regions. We use Element's ability to generate paired end sequencing with longer insert sizes than typical short-read sequencing. We show that longer insert sizes result in even higher accuracy, with long insert Element sequencing giving more accurate genome analyses at all coverages.</p><p><strong>Conclusions: </strong>New options for sequencing technologies can analyze genomes comparably or better than prior standard methods.</p>","PeriodicalId":8958,"journal":{"name":"BMC Bioinformatics","volume":"26 1","pages":"194"},"PeriodicalIF":3.3000,"publicationDate":"2025-07-25","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12291380/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"BMC Bioinformatics","FirstCategoryId":"99","ListUrlMain":"https://doi.org/10.1186/s12859-025-06191-4","RegionNum":3,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"BIOCHEMICAL RESEARCH METHODS","Score":null,"Total":0}
引用次数: 0
Abstract
Background: New sequencing technologies provide options for the scientific community to design studies and build clinical workflows. These options expand user choice, and can enable more accurate, scalable, or affordable workflows depending on the fit between scientist needs and platform capability. However, it is essential to understand the performance of these new technologies for different tasks, especially for capabilities that were not possible or tractable in prior technologies. We investigate the new sequencing technology avidity from Element Biosciences. to help the scientific community understand the performance of the options to generate sequencing data.
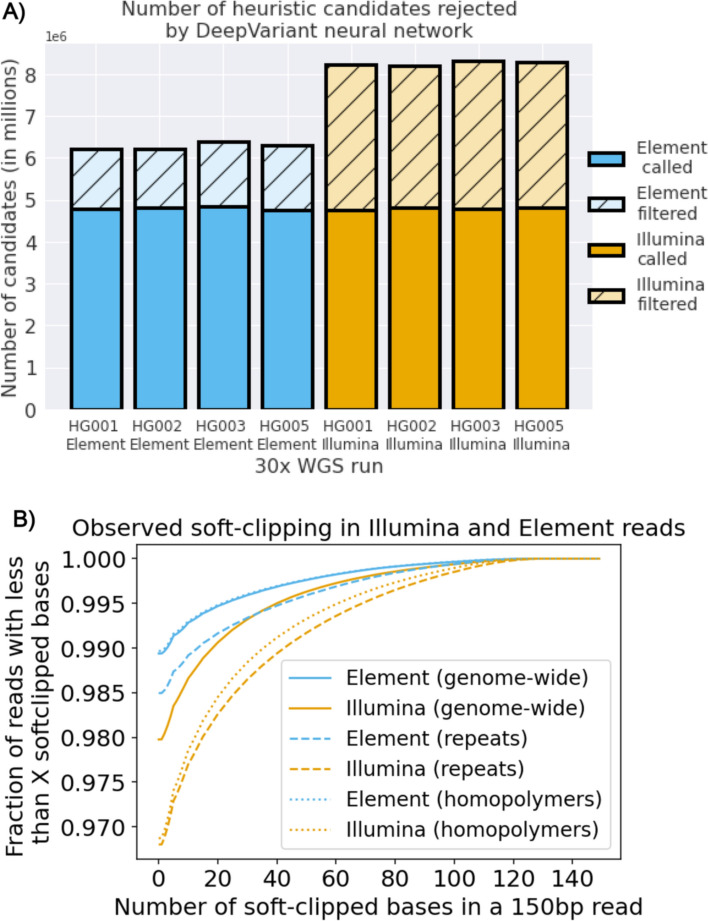
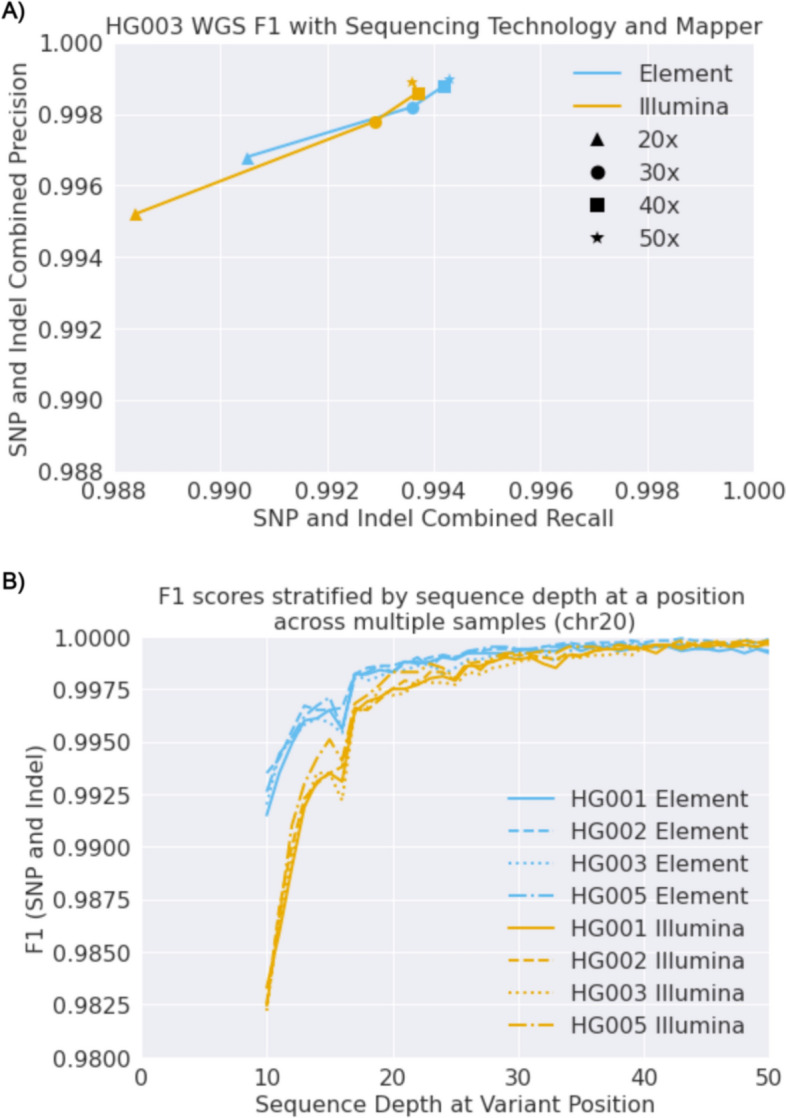
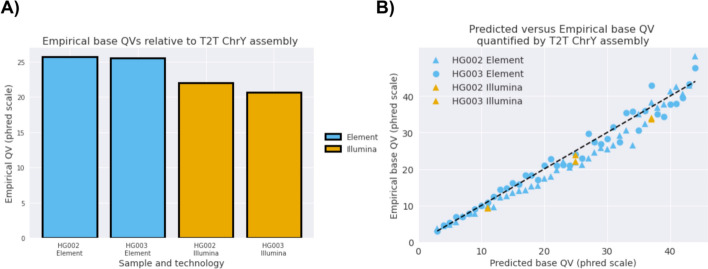
Results: We show that Element whole genome sequencing achieves higher mapping and variant calling accuracy compared to Illumina sequencing at the same coverage, with larger differences at lower coverages (20-30x). We quantify base error rates of Element reads, finding lower error rates, especially in homopolymer and tandem repeat regions. We use Element's ability to generate paired end sequencing with longer insert sizes than typical short-read sequencing. We show that longer insert sizes result in even higher accuracy, with long insert Element sequencing giving more accurate genome analyses at all coverages.
Conclusions: New options for sequencing technologies can analyze genomes comparably or better than prior standard methods.
期刊介绍:
BMC Bioinformatics is an open access, peer-reviewed journal that considers articles on all aspects of the development, testing and novel application of computational and statistical methods for the modeling and analysis of all kinds of biological data, as well as other areas of computational biology.
BMC Bioinformatics is part of the BMC series which publishes subject-specific journals focused on the needs of individual research communities across all areas of biology and medicine. We offer an efficient, fair and friendly peer review service, and are committed to publishing all sound science, provided that there is some advance in knowledge presented by the work.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


