{"title":"Hybrid representation learning for human m<sup>6</sup>A modifications with chromosome-level generalizability.","authors":"Muhammad Tahir, Sheela Ramanna, Qian Liu","doi":"10.1093/bioadv/vbaf170","DOIUrl":null,"url":null,"abstract":"<p><strong>Motivation: </strong><math> <mrow> <mrow> <msup><mrow><mi>N</mi></mrow> <mn>6</mn></msup> </mrow> <mo>-</mo> <mtext>methyladenosine</mtext></mrow> </math> ( <math> <mrow> <mrow> <msup><mrow><mi>m</mi></mrow> <mn>6</mn></msup> </mrow> <mi>A</mi></mrow> </math> ) is the most abundant internal modification in eukaryotic mRNA and plays essential roles in post-transcriptional gene regulation. While several deep learning approaches have been proposed to predict <math> <mrow> <mrow> <msup><mrow><mi>m</mi></mrow> <mn>6</mn></msup> </mrow> <mi>A</mi></mrow> </math> sites, most suffer from limited chromosome-level generalizability due to evaluation on randomly split datasets.</p><p><strong>Results: </strong>In this study, we propose two novel hybrid deep learning models-Hybrid Model and Hybrid Deep Model-that integrate local sequence features (<i>k</i>-mers) and contextual embeddings via convolutional neural networks to improve predictive performance and generalization. We evaluate these models using both a Random-Split strategy and a more biologically realistic Leave-One-Chromosome-Out setting to ensure robustness across genomic regions. Our proposed models outperform the state-of-the-art m6A-TCPred model across all key evaluation metrics. Hybrid Deep Model achieves the highest accuracy under Random-Split, while Hybrid Model demonstrates superior generalization under Leave-One-Chromosome-Out, indicating that deep global representations may overfit in chromosome-independent settings. These findings underscore the importance of rigorous validation strategies and offer insights into designing robust <math> <mrow> <mrow> <msup><mrow><mi>m</mi></mrow> <mn>6</mn></msup> </mrow> <mi>A</mi></mrow> </math> predictors.</p><p><strong>Availability and implementation: </strong>Source code and datasets are available at: https://github.com/malikmtahir/LOCO-m6A.</p>","PeriodicalId":72368,"journal":{"name":"Bioinformatics advances","volume":"5 1","pages":"vbaf170"},"PeriodicalIF":2.8000,"publicationDate":"2025-07-14","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12288952/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Bioinformatics advances","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1093/bioadv/vbaf170","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2025/1/1 0:00:00","PubModel":"eCollection","JCR":"Q2","JCRName":"MATHEMATICAL & COMPUTATIONAL BIOLOGY","Score":null,"Total":0}
引用次数: 0
Abstract
Motivation: ( ) is the most abundant internal modification in eukaryotic mRNA and plays essential roles in post-transcriptional gene regulation. While several deep learning approaches have been proposed to predict sites, most suffer from limited chromosome-level generalizability due to evaluation on randomly split datasets.
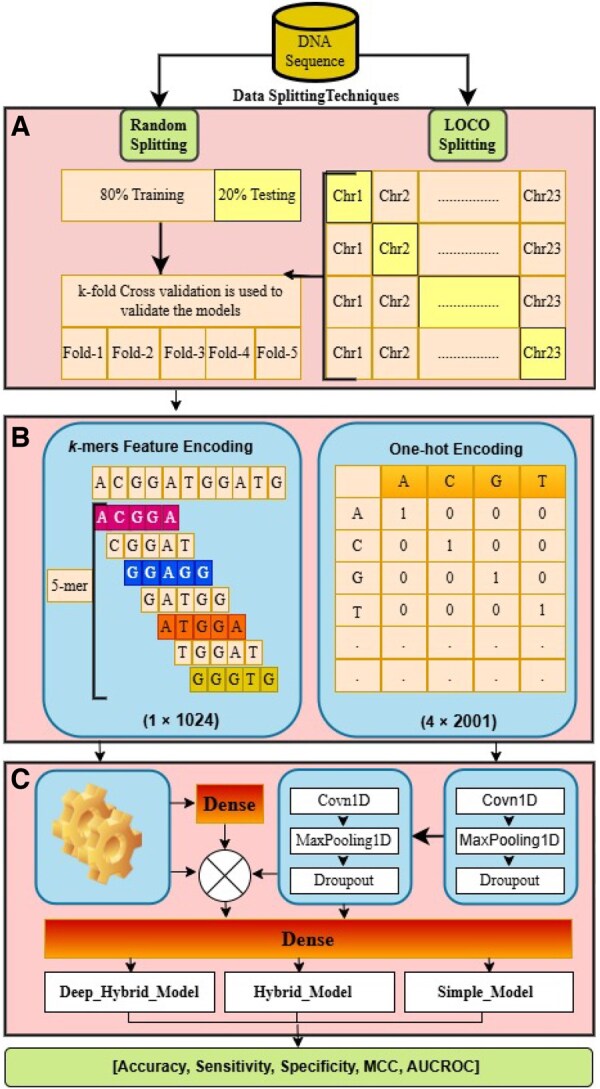
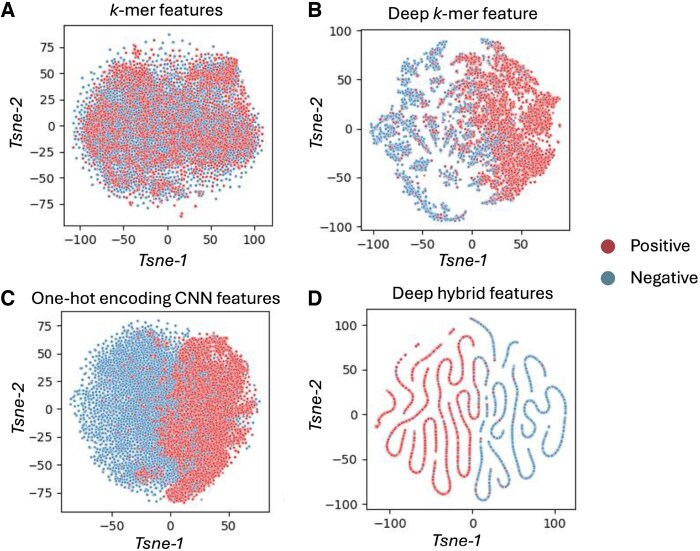
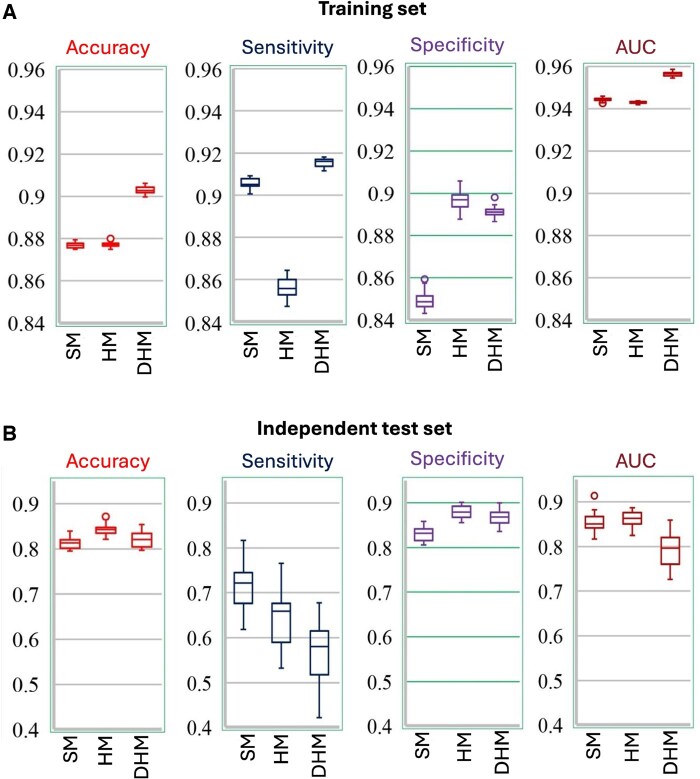
Results: In this study, we propose two novel hybrid deep learning models-Hybrid Model and Hybrid Deep Model-that integrate local sequence features (k-mers) and contextual embeddings via convolutional neural networks to improve predictive performance and generalization. We evaluate these models using both a Random-Split strategy and a more biologically realistic Leave-One-Chromosome-Out setting to ensure robustness across genomic regions. Our proposed models outperform the state-of-the-art m6A-TCPred model across all key evaluation metrics. Hybrid Deep Model achieves the highest accuracy under Random-Split, while Hybrid Model demonstrates superior generalization under Leave-One-Chromosome-Out, indicating that deep global representations may overfit in chromosome-independent settings. These findings underscore the importance of rigorous validation strategies and offer insights into designing robust predictors.
Availability and implementation: Source code and datasets are available at: https://github.com/malikmtahir/LOCO-m6A.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


