Alayne P Meyer, Daniel C Koboldt, Swetha Ramadesikan, Kristin Zajo, Maria E Hernandez Gonzalez, Anthony R Miller, Douglas Depoorter, Catherine P Comer, James I Geller, Katherine Somers, Nilay Shah, Marco L Leung
{"title":"Three Siblings With an Attenuated Presentation of Perlman Syndrome: A Case Report and Literature Review.","authors":"Alayne P Meyer, Daniel C Koboldt, Swetha Ramadesikan, Kristin Zajo, Maria E Hernandez Gonzalez, Anthony R Miller, Douglas Depoorter, Catherine P Comer, James I Geller, Katherine Somers, Nilay Shah, Marco L Leung","doi":"10.1002/mgg3.70124","DOIUrl":null,"url":null,"abstract":"<p><strong>Introduction: </strong>Perlman syndrome is a rare autosomal recessive overgrowth disorder with a predisposition to Wilms tumor, caused by biallelic variants in DIS3L2. The majority of patients die in infancy due to respiratory and/or renal failure, limiting the reports of patients surviving into childhood.</p><p><strong>Methods: </strong>Exome sequencing was performed in the proband and her older brother. A younger sibling subsequently underwent targeted variant analysis. RNA sequencing was utilized to investigate the functional impact of the missense variant.</p><p><strong>Results: </strong>Three siblings presented at birth with fetal macrosomia, dysmorphic facial features, and facial hypotonia. The proband had early speech delay and was diagnosed with Wilms tumor at 3 years old. Her brothers both had developmental delay presenting within the first year of life. Genetic testing identified compound heterozygous variants in DIS3L2 (NM_152383.5): c.127C>T (p.Arg43Ter) (paternal)/c.2381G>A (p.Arg794His) (maternal).</p><p><strong>Conclusion: </strong>Our findings expand the genetic and clinical spectrums associated with Perlman syndrome and increase the understanding of the phenotype observed in childhood. They also support consideration of genetic testing for Perlman syndrome in individuals and sibships with macrosomia, developmental delay, and characteristic facial dysmorphisms, with or without the presence of Wilms tumor.</p>","PeriodicalId":18852,"journal":{"name":"Molecular Genetics & Genomic Medicine","volume":"13 7","pages":"e70124"},"PeriodicalIF":1.6000,"publicationDate":"2025-07-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12288098/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Molecular Genetics & Genomic Medicine","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1002/mgg3.70124","RegionNum":4,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q4","JCRName":"GENETICS & HEREDITY","Score":null,"Total":0}
引用次数: 0
Abstract
Introduction: Perlman syndrome is a rare autosomal recessive overgrowth disorder with a predisposition to Wilms tumor, caused by biallelic variants in DIS3L2. The majority of patients die in infancy due to respiratory and/or renal failure, limiting the reports of patients surviving into childhood.
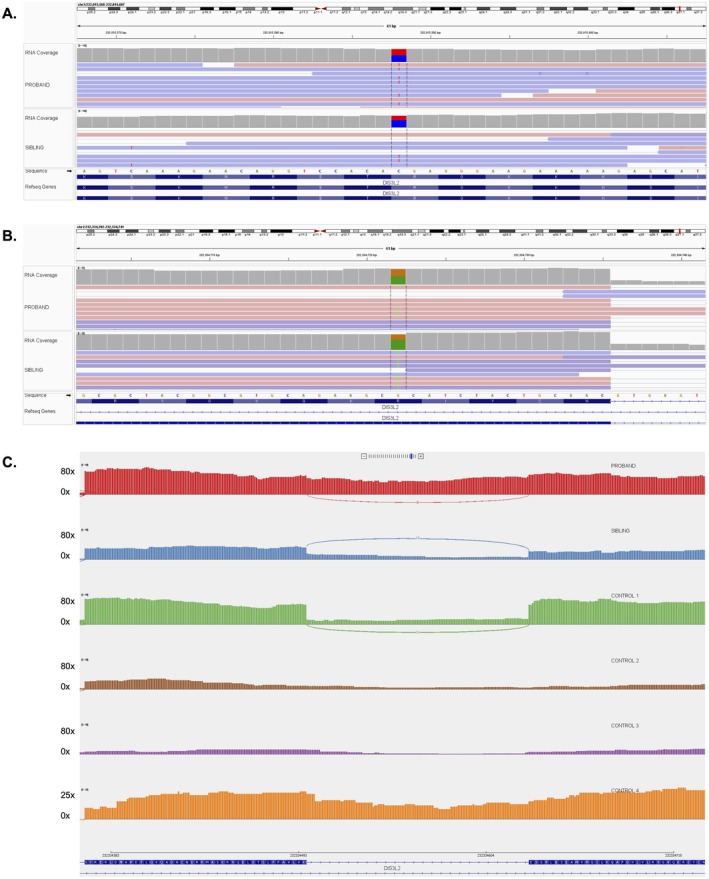
Methods: Exome sequencing was performed in the proband and her older brother. A younger sibling subsequently underwent targeted variant analysis. RNA sequencing was utilized to investigate the functional impact of the missense variant.
Results: Three siblings presented at birth with fetal macrosomia, dysmorphic facial features, and facial hypotonia. The proband had early speech delay and was diagnosed with Wilms tumor at 3 years old. Her brothers both had developmental delay presenting within the first year of life. Genetic testing identified compound heterozygous variants in DIS3L2 (NM_152383.5): c.127C>T (p.Arg43Ter) (paternal)/c.2381G>A (p.Arg794His) (maternal).
Conclusion: Our findings expand the genetic and clinical spectrums associated with Perlman syndrome and increase the understanding of the phenotype observed in childhood. They also support consideration of genetic testing for Perlman syndrome in individuals and sibships with macrosomia, developmental delay, and characteristic facial dysmorphisms, with or without the presence of Wilms tumor.
期刊介绍:
Molecular Genetics & Genomic Medicine is a peer-reviewed journal for rapid dissemination of quality research related to the dynamically developing areas of human, molecular and medical genetics. The journal publishes original research articles covering findings in phenotypic, molecular, biological, and genomic aspects of genomic variation, inherited disorders and birth defects. The broad publishing spectrum of Molecular Genetics & Genomic Medicine includes rare and common disorders from diagnosis to treatment. Examples of appropriate articles include reports of novel disease genes, functional studies of genetic variants, in-depth genotype-phenotype studies, genomic analysis of inherited disorders, molecular diagnostic methods, medical bioinformatics, ethical, legal, and social implications (ELSI), and approaches to clinical diagnosis. Molecular Genetics & Genomic Medicine provides a scientific home for next generation sequencing studies of rare and common disorders, which will make research in this fascinating area easily and rapidly accessible to the scientific community. This will serve as the basis for translating next generation sequencing studies into individualized diagnostics and therapeutics, for day-to-day medical care.
Molecular Genetics & Genomic Medicine publishes original research articles, reviews, and research methods papers, along with invited editorials and commentaries. Original research papers must report well-conducted research with conclusions supported by the data presented.



 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


