{"title":"LC-QTOF-MS<sup>E</sup> with MS<sup>1</sup>-based precursor ion quantification and SiMD-assisted identification enhances human urine metabolite analysis.","authors":"Alongkorn Kurilung, Suphitcha Limjiasahapong, Kwanjeera Wanichthanarak, Weerawan Manokasemsan, Khwanta Kaewnarin, Kassaporn Duangkumpha, Siriphan Manocheewa, Rossarin Tansawat, Roongruedee Chaiteerakij, Intawat Nookaew, Yongyut Sirivatanauksorn, Sakda Khoomrung","doi":"10.1016/j.csbj.2025.07.009","DOIUrl":null,"url":null,"abstract":"<p><p>This study presents the development and validation of a liquid chromatography-quadrupole-time-of-flight mass spectrometry method with data-independent acquisition (LC-QTOF-MS<sup>E</sup>) for targeted quantification, post-targeted screening, and untargeted metabolite profiling. Using MS<sup>1</sup>-based precursor ion quantification, the method demonstrated excellent analytical performance with linearity (<i>R</i>² > 0.99), accuracy (84 %-131 %), and precision (1 %-17 % relative standard deviation (RSD)). Although LC-QTOF‑MS<sup>E</sup> sensitivity is at least nine-fold lower than LC-triple quadrupole MS with multiple reaction monitoring, it remains adequate for quantifying urinary metabolites, particularly those that fragment poorly or yield low‑intensity product ions. For post‑targeted screening and untargeted profiling, an in‑house reference library (the Siriraj Metabolomics Data Warehouse, SiMD), comprising 174 curated metabolite standards, was integrated into the workflow to enhance metabolite identification confidence. The official website for SiMD can be accessed at https://si-simd.com/. To demonstrate the method's utility, 11 amino and organic acids were quantified in urine samples from 100 healthy individuals. Four compounds-L-methionine, L-histidine, L-tryptophan, and <i>trans</i>-ferulic acid-were significantly higher levels in females (<i>P</i> < 0.05), likely reflecting sex-specific physiological or dietary intake differences. Post‑targeted screening identified 29 additional metabolites and assigned them to level 1 (<i>m</i>/<i>z</i>, RT, isotope pattern, and MS/MS spectra matched to reference standards) based on the Metabolomics Standards Initiative guidelines. Untargeted retrospective profiling revealed level 1 seven metabolites, including ribitol, creatine, glucuronic acid, <i>trans</i>-ferulic acid, succinic acid, dimethylglycine, and 3-hydroxyphenylacetic acid related to sex variation (VIP > 1.5). In summary, the LC-QTOF-MS<sup>E</sup> method coupled with SiMD provides a robust and comprehensive workflow for metabolomics analysis. It enables reliable target quantification and enhances confidence in metabolite identification while also reducing sample and instrumental demands. These features make it particularly well-suited for clinical metabolomics studies.</p>","PeriodicalId":10715,"journal":{"name":"Computational and structural biotechnology journal","volume":"27 ","pages":"3079-3089"},"PeriodicalIF":4.1000,"publicationDate":"2025-07-10","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12284563/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Computational and structural biotechnology journal","FirstCategoryId":"99","ListUrlMain":"https://doi.org/10.1016/j.csbj.2025.07.009","RegionNum":2,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2025/1/1 0:00:00","PubModel":"eCollection","JCR":"Q2","JCRName":"BIOCHEMISTRY & MOLECULAR BIOLOGY","Score":null,"Total":0}
引用次数: 0
Abstract
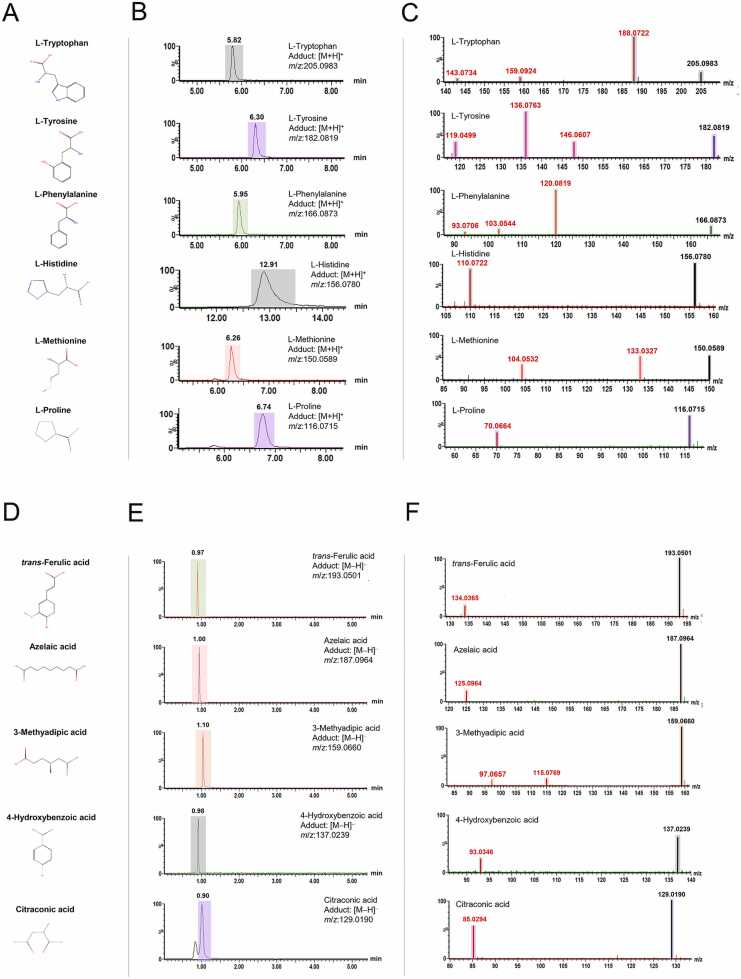
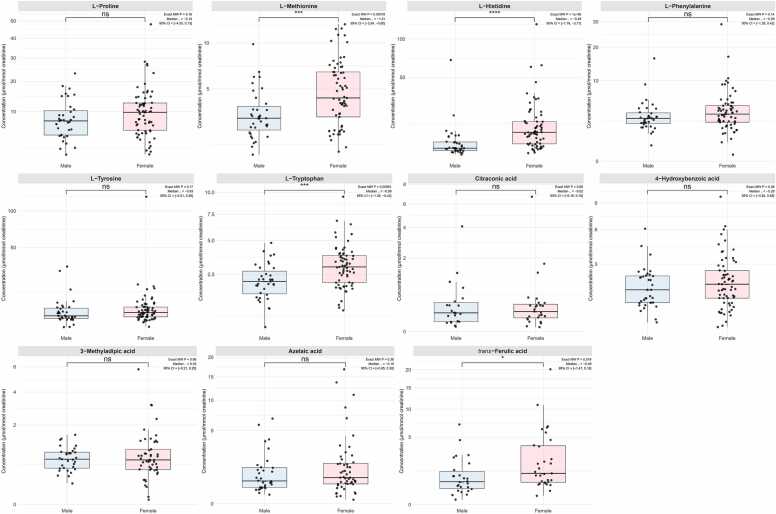
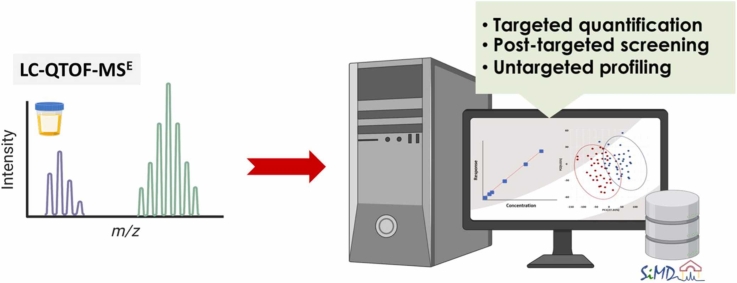
This study presents the development and validation of a liquid chromatography-quadrupole-time-of-flight mass spectrometry method with data-independent acquisition (LC-QTOF-MSE) for targeted quantification, post-targeted screening, and untargeted metabolite profiling. Using MS1-based precursor ion quantification, the method demonstrated excellent analytical performance with linearity (R² > 0.99), accuracy (84 %-131 %), and precision (1 %-17 % relative standard deviation (RSD)). Although LC-QTOF‑MSE sensitivity is at least nine-fold lower than LC-triple quadrupole MS with multiple reaction monitoring, it remains adequate for quantifying urinary metabolites, particularly those that fragment poorly or yield low‑intensity product ions. For post‑targeted screening and untargeted profiling, an in‑house reference library (the Siriraj Metabolomics Data Warehouse, SiMD), comprising 174 curated metabolite standards, was integrated into the workflow to enhance metabolite identification confidence. The official website for SiMD can be accessed at https://si-simd.com/. To demonstrate the method's utility, 11 amino and organic acids were quantified in urine samples from 100 healthy individuals. Four compounds-L-methionine, L-histidine, L-tryptophan, and trans-ferulic acid-were significantly higher levels in females (P < 0.05), likely reflecting sex-specific physiological or dietary intake differences. Post‑targeted screening identified 29 additional metabolites and assigned them to level 1 (m/z, RT, isotope pattern, and MS/MS spectra matched to reference standards) based on the Metabolomics Standards Initiative guidelines. Untargeted retrospective profiling revealed level 1 seven metabolites, including ribitol, creatine, glucuronic acid, trans-ferulic acid, succinic acid, dimethylglycine, and 3-hydroxyphenylacetic acid related to sex variation (VIP > 1.5). In summary, the LC-QTOF-MSE method coupled with SiMD provides a robust and comprehensive workflow for metabolomics analysis. It enables reliable target quantification and enhances confidence in metabolite identification while also reducing sample and instrumental demands. These features make it particularly well-suited for clinical metabolomics studies.
期刊介绍:
Computational and Structural Biotechnology Journal (CSBJ) is an online gold open access journal publishing research articles and reviews after full peer review. All articles are published, without barriers to access, immediately upon acceptance. The journal places a strong emphasis on functional and mechanistic understanding of how molecular components in a biological process work together through the application of computational methods. Structural data may provide such insights, but they are not a pre-requisite for publication in the journal. Specific areas of interest include, but are not limited to:
Structure and function of proteins, nucleic acids and other macromolecules
Structure and function of multi-component complexes
Protein folding, processing and degradation
Enzymology
Computational and structural studies of plant systems
Microbial Informatics
Genomics
Proteomics
Metabolomics
Algorithms and Hypothesis in Bioinformatics
Mathematical and Theoretical Biology
Computational Chemistry and Drug Discovery
Microscopy and Molecular Imaging
Nanotechnology
Systems and Synthetic Biology

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


