Lina M. Bolivar-Pineda, Elena V. Basiuk, Vladimir A. Basiuk
{"title":"Noncovalent dyads of lanthanide nitride cluster fullerenes Ln3N@C80 and bisphthalocyanines LnPc2: Insights from DFT calculations","authors":"Lina M. Bolivar-Pineda, Elena V. Basiuk, Vladimir A. Basiuk","doi":"10.1007/s00894-025-06415-7","DOIUrl":null,"url":null,"abstract":"<div><h3>Context</h3><p>Lanthanide-based systems, such as nitride cluster fullerenes Ln<sub>3</sub>N@C<sub>80</sub> and bipthalocyanines LnPc<sub>2</sub> (Pc = phthalocyanine ligand), are of interest for their magnetic, fluorescent and electronic properties. In this regard, we performed DFT characterization to investigate the changes in structure and electronic properties for noncovalently interacting lanthanide (Ln; where Ln = La, Ce, Gd and Lu) nitride cluster fullerenes and bisphthalocyanines to form Ln<sub>3</sub>N@C<sub>80</sub> + LnPc<sub>2</sub> dyads. The optimized geometries, formation and frontier orbital energies, HOMO-LUMO plots, charge and spin of Ln and N(NCF) atoms, as well as spin density plots of the dyads were analyzed in comparison with those of isolated Ln<sub>3</sub>N@C<sub>80</sub> and LnPc<sub>2</sub> components. In addition to LnPc<sub>2</sub> bending distortion, the noncovalent dyad formation alters the geometry of the encapsulated Ln<sub>3</sub>N cluster, favoring more planar or pyramidal geometries, depending on the case. The HOMO and LUMO orbitals are found on bisphthalocyanines, being localized on the isoindole units, except for Ce<sub>3</sub>N@C<sub>80</sub> + CePc<sub>2</sub> dyad, where the LUMO was found on the central metal of CePc<sub>2</sub>. The HOMO-LUMO gap energy is lower for the dyads compared to isolated NCFs, being rather close to the gap energy of bisphthalocyanines. The changes in spin density distribution are evident in the dyads containing Ce and Gd atoms, contrary to their La and Lu-derived counterparts. The interaction of Ce<sub>3</sub>N@C<sub>80</sub> and Gd<sub>3</sub>N@C<sub>80</sub> with CePc<sub>2</sub> and GdPc<sub>2</sub>, respectively, causes redistribution of the spin density, with changes in the orientation of spin-up and spin-down electrons in the encapsulated Ce<sub>3</sub>N and Gd<sub>3</sub>N clusters.</p><h3>Methods</h3><p>The geometry optimization and electronic properties calculations based on density functional theory were performed using the DMol<sup>3</sup> module of Material Studio 8.0 software package from Accelrys Inc. The computational parameters selected included the general gradient approximation functional PBE, combined with a long-range dispersion correction developed by Grimme (PBE-D2), the double numerical basis set (DN), equivalent to the 6-31G Pople-type basis set along with the DFT semiconductor pseudopotentials. To mitigate the self-consistent field convergence problems, the thermal smearing technique was applied, with a final very small value of 0.0001 Ha (equivalent to 31.6 K temperature), or Fermi orbital occupancy in some cases.</p><h3>Graphical Abstract</h3>\n<div><figure><div><div><picture><source><img></source></picture></div></div></figure></div></div>","PeriodicalId":651,"journal":{"name":"Journal of Molecular Modeling","volume":"31 8","pages":""},"PeriodicalIF":2.5000,"publicationDate":"2025-07-24","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12287152/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Molecular Modeling","FirstCategoryId":"92","ListUrlMain":"https://link.springer.com/article/10.1007/s00894-025-06415-7","RegionNum":4,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q4","JCRName":"BIOCHEMISTRY & MOLECULAR BIOLOGY","Score":null,"Total":0}
引用次数: 0
Abstract
Context
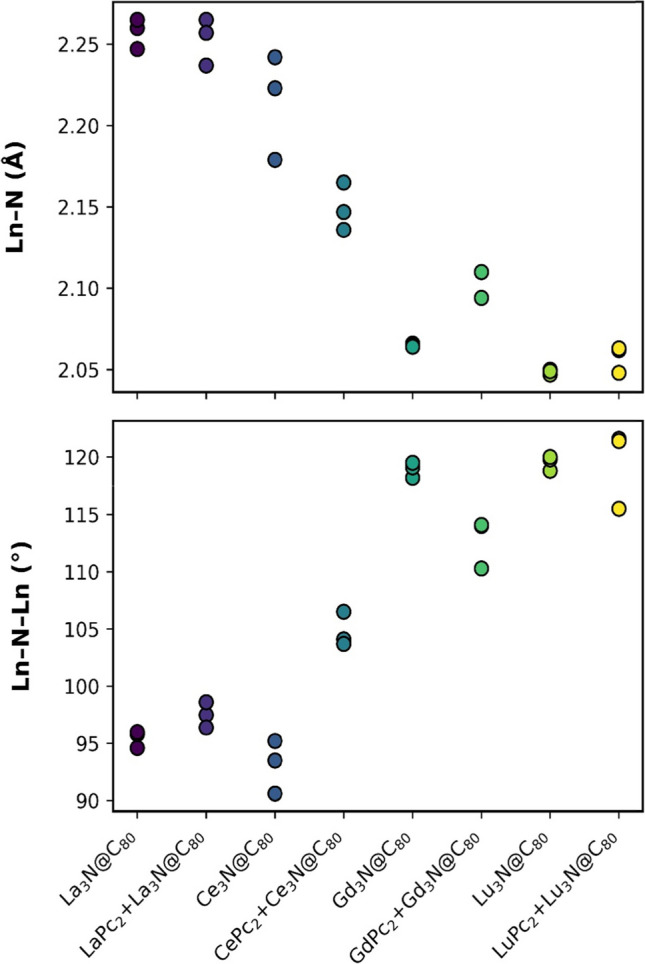
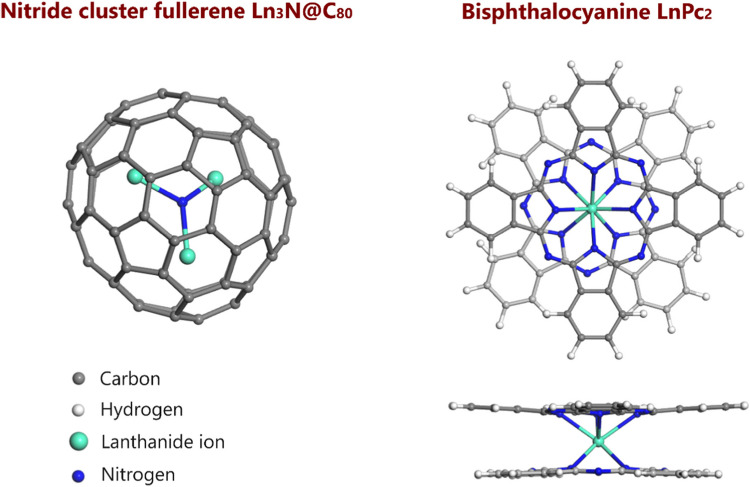
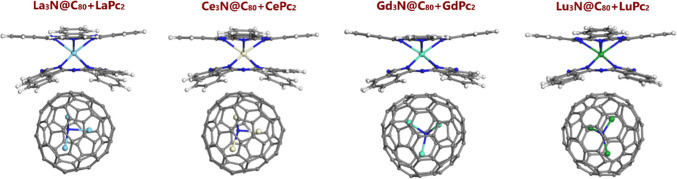
Lanthanide-based systems, such as nitride cluster fullerenes Ln3N@C80 and bipthalocyanines LnPc2 (Pc = phthalocyanine ligand), are of interest for their magnetic, fluorescent and electronic properties. In this regard, we performed DFT characterization to investigate the changes in structure and electronic properties for noncovalently interacting lanthanide (Ln; where Ln = La, Ce, Gd and Lu) nitride cluster fullerenes and bisphthalocyanines to form Ln3N@C80 + LnPc2 dyads. The optimized geometries, formation and frontier orbital energies, HOMO-LUMO plots, charge and spin of Ln and N(NCF) atoms, as well as spin density plots of the dyads were analyzed in comparison with those of isolated Ln3N@C80 and LnPc2 components. In addition to LnPc2 bending distortion, the noncovalent dyad formation alters the geometry of the encapsulated Ln3N cluster, favoring more planar or pyramidal geometries, depending on the case. The HOMO and LUMO orbitals are found on bisphthalocyanines, being localized on the isoindole units, except for Ce3N@C80 + CePc2 dyad, where the LUMO was found on the central metal of CePc2. The HOMO-LUMO gap energy is lower for the dyads compared to isolated NCFs, being rather close to the gap energy of bisphthalocyanines. The changes in spin density distribution are evident in the dyads containing Ce and Gd atoms, contrary to their La and Lu-derived counterparts. The interaction of Ce3N@C80 and Gd3N@C80 with CePc2 and GdPc2, respectively, causes redistribution of the spin density, with changes in the orientation of spin-up and spin-down electrons in the encapsulated Ce3N and Gd3N clusters.
Methods
The geometry optimization and electronic properties calculations based on density functional theory were performed using the DMol3 module of Material Studio 8.0 software package from Accelrys Inc. The computational parameters selected included the general gradient approximation functional PBE, combined with a long-range dispersion correction developed by Grimme (PBE-D2), the double numerical basis set (DN), equivalent to the 6-31G Pople-type basis set along with the DFT semiconductor pseudopotentials. To mitigate the self-consistent field convergence problems, the thermal smearing technique was applied, with a final very small value of 0.0001 Ha (equivalent to 31.6 K temperature), or Fermi orbital occupancy in some cases.
期刊介绍:
The Journal of Molecular Modeling focuses on "hardcore" modeling, publishing high-quality research and reports. Founded in 1995 as a purely electronic journal, it has adapted its format to include a full-color print edition, and adjusted its aims and scope fit the fast-changing field of molecular modeling, with a particular focus on three-dimensional modeling.
Today, the journal covers all aspects of molecular modeling including life science modeling; materials modeling; new methods; and computational chemistry.
Topics include computer-aided molecular design; rational drug design, de novo ligand design, receptor modeling and docking; cheminformatics, data analysis, visualization and mining; computational medicinal chemistry; homology modeling; simulation of peptides, DNA and other biopolymers; quantitative structure-activity relationships (QSAR) and ADME-modeling; modeling of biological reaction mechanisms; and combined experimental and computational studies in which calculations play a major role.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


