Rei Oshima, Mikito Fujinami, Yuya Nakajima, Hiromi Nakai
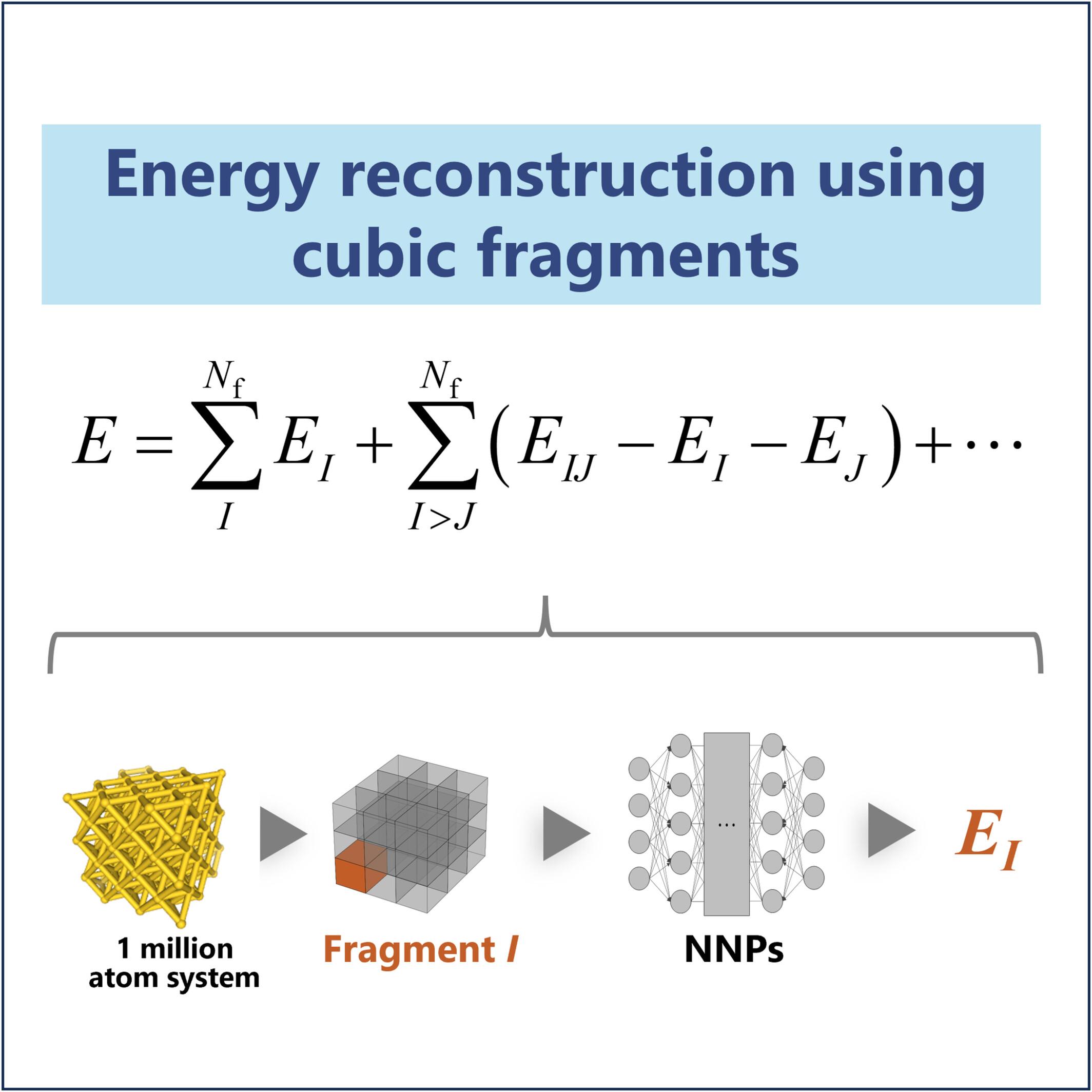
{"title":"Large-Scale Calculations by Integrating the Fragmentation Approach With Neural Network Potentials","authors":"Rei Oshima, Mikito Fujinami, Yuya Nakajima, Hiromi Nakai","doi":"10.1002/jcc.70193","DOIUrl":null,"url":null,"abstract":"<div>\n \n <p>A fragmentation method is introduced to enable large-scale molecular simulations using neural network potentials (NNPs). The method partitions a system into cube-shaped fragments and reconstructs the total energy using a many-body expansion formalism with a distance-based cut-off approximation. Validation with Au, NaCl, diamond, H<sub>2</sub>O, and graphite crystals demonstrated that including three-body interactions with 26 neighboring fragments reduces per-atom energy error to within 0.04 eV. This approach enables simulations of systems with up to 1 million atoms, surpassing conventional NNP limits. The scaling exponent for three-body calculations remains below 1.64, suggesting feasibility for even larger-scale applications.</p>\n </div>","PeriodicalId":188,"journal":{"name":"Journal of Computational Chemistry","volume":"46 20","pages":""},"PeriodicalIF":4.8000,"publicationDate":"2025-07-24","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Computational Chemistry","FirstCategoryId":"92","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1002/jcc.70193","RegionNum":3,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"CHEMISTRY, MULTIDISCIPLINARY","Score":null,"Total":0}
引用次数: 0
Abstract
A fragmentation method is introduced to enable large-scale molecular simulations using neural network potentials (NNPs). The method partitions a system into cube-shaped fragments and reconstructs the total energy using a many-body expansion formalism with a distance-based cut-off approximation. Validation with Au, NaCl, diamond, H2O, and graphite crystals demonstrated that including three-body interactions with 26 neighboring fragments reduces per-atom energy error to within 0.04 eV. This approach enables simulations of systems with up to 1 million atoms, surpassing conventional NNP limits. The scaling exponent for three-body calculations remains below 1.64, suggesting feasibility for even larger-scale applications.
期刊介绍:
This distinguished journal publishes articles concerned with all aspects of computational chemistry: analytical, biological, inorganic, organic, physical, and materials. The Journal of Computational Chemistry presents original research, contemporary developments in theory and methodology, and state-of-the-art applications. Computational areas that are featured in the journal include ab initio and semiempirical quantum mechanics, density functional theory, molecular mechanics, molecular dynamics, statistical mechanics, cheminformatics, biomolecular structure prediction, molecular design, and bioinformatics.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


