{"title":"Rapid Assessment of Virtually Synthesizable Chemical Structures via Support Vector Machine Models.","authors":"Yuto Iwasaki, Tomoyuki Miyao","doi":"10.1002/minf.70000","DOIUrl":null,"url":null,"abstract":"<p><p>Support vector machine (SVM) and support vector regression (SVR) are widely used for building quantitative structure-activity relationship models for small- and medium-sized datasets. Although SVM and SVR models can efficiently predict compound activity, evaluating billions of molecules remains challenging, which sometimes occurs when screening the virtual molecules derived through virtual synthesis. Herein, we present an SVM-/SVR-based method for screening virtually synthesizable molecules based on their reactants. The proposed method employs a combination of reactant-wise kernel functions for fast evaluation without sacrificing prediction accuracy. Tested on 120 small molecular activity datasets against 10 macromolecule targets, the proposed SVR models with data augmentation worked equally to standard SVR models with the Tanimoto kernel. As a demonstration, exhaustive 6.4 × 10<sup>12</sup> reactant combinations were evaluated by an SVR model within 8 days on a single desktop computer, enabling large-scale screening without sampling.</p>","PeriodicalId":18853,"journal":{"name":"Molecular Informatics","volume":"44 7","pages":"e202500039"},"PeriodicalIF":3.1000,"publicationDate":"2025-07-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12278806/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Molecular Informatics","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1002/minf.70000","RegionNum":4,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"CHEMISTRY, MEDICINAL","Score":null,"Total":0}
引用次数: 0
Abstract
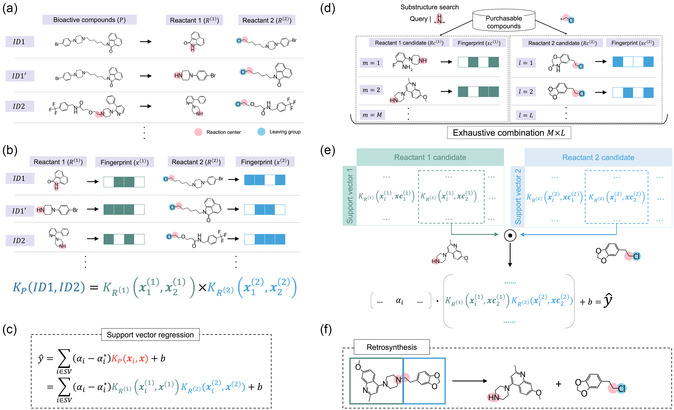
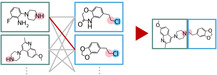
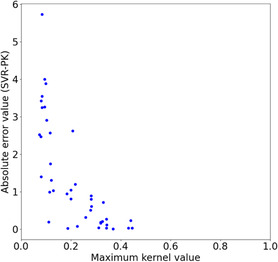
Support vector machine (SVM) and support vector regression (SVR) are widely used for building quantitative structure-activity relationship models for small- and medium-sized datasets. Although SVM and SVR models can efficiently predict compound activity, evaluating billions of molecules remains challenging, which sometimes occurs when screening the virtual molecules derived through virtual synthesis. Herein, we present an SVM-/SVR-based method for screening virtually synthesizable molecules based on their reactants. The proposed method employs a combination of reactant-wise kernel functions for fast evaluation without sacrificing prediction accuracy. Tested on 120 small molecular activity datasets against 10 macromolecule targets, the proposed SVR models with data augmentation worked equally to standard SVR models with the Tanimoto kernel. As a demonstration, exhaustive 6.4 × 1012 reactant combinations were evaluated by an SVR model within 8 days on a single desktop computer, enabling large-scale screening without sampling.
期刊介绍:
Molecular Informatics is a peer-reviewed, international forum for publication of high-quality, interdisciplinary research on all molecular aspects of bio/cheminformatics and computer-assisted molecular design. Molecular Informatics succeeded QSAR & Combinatorial Science in 2010.
Molecular Informatics presents methodological innovations that will lead to a deeper understanding of ligand-receptor interactions, macromolecular complexes, molecular networks, design concepts and processes that demonstrate how ideas and design concepts lead to molecules with a desired structure or function, preferably including experimental validation.
The journal''s scope includes but is not limited to the fields of drug discovery and chemical biology, protein and nucleic acid engineering and design, the design of nanomolecular structures, strategies for modeling of macromolecular assemblies, molecular networks and systems, pharmaco- and chemogenomics, computer-assisted screening strategies, as well as novel technologies for the de novo design of biologically active molecules. As a unique feature Molecular Informatics publishes so-called "Methods Corner" review-type articles which feature important technological concepts and advances within the scope of the journal.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


