mCNN-GenEfflux: enhanced predicting Efflux protein and their super families by using generative proteins combined with multiple windows convolution neural networks
IF 3.1
4区 生物学
Q2 BIOLOGY
引用次数: 0
Abstract
Efflux transporters play a critical role in bacterial antibiotic resistance by facilitating the removal of harmful substances. These are classified into five distinct families: ABC, MFS, MATE, RND, and SMR. The significant sequence variability among these families, coupled with insufficient functional annotation, presents considerable challenges for traditional categorization methods. We hypothesize that integrating of ProtGPT2-generated efflux protein sequences with a multi-window convolutional neural network (mCNN) is proposed to enhance classification accuracy by effectively capturing local motifs and broader evolutionary patterns often overlooked by previous methods. Generative models like ProtGPT2 are effective for this purpose, as they generate a variety of sequence variants that reflect patterns observed in natural efflux families, thereby minimizing issues related to data scarcity. The proposed GenEfflux framework, unlike alignment-based methods such as HHblits and single-feature CNNs, combines generative sequence expanding with multi-scale evolutionary feature extraction via Position-Specific Scoring Matrices (PSSMs), thereby enhancing the understanding of sequence-function relationships. In comparative evaluations, GenEfflux consistently outperformed the baseline deepEfflux model across all Efflux transporter classes. In Class B, sensitivity increased from 0.5385 to 0.9999, and the Matthews correlation coefficient (MCC) rose from 0.4397 to 0.9327. In Class C, accuracy improved from 0.8977 to 0.9668, alongside an increase in MCC from 0.7668 to 0.9331. The findings demonstrate that sequences generated by ProtGPT2 possess functional relevance and improve classification effectiveness. GenEfflux suggestions a comprehensive framework for enhancing efflux protein analysis and advancing research on antibiotic resistance.
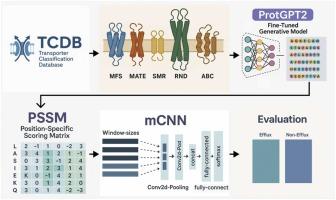
mCNN-GenEfflux:利用生成蛋白结合多窗口卷积神经网络增强对外排蛋白及其超家族的预测
外排转运体通过促进有害物质的清除在细菌抗生素耐药性中起关键作用。它们被分为五个不同的家族:ABC、MFS、MATE、RND和SMR。这些科之间的序列差异显著,加上功能注释不足,对传统的分类方法提出了相当大的挑战。我们假设将protgpt2产生的外排蛋白序列与多窗口卷积神经网络(mCNN)整合,通过有效捕获局部基序和更广泛的进化模式来提高分类精度,这些模式通常被以前的方法所忽视。像ProtGPT2这样的生成模型对于这一目的是有效的,因为它们生成了各种序列变体,反映了在自然外流家族中观察到的模式,从而最大限度地减少了与数据稀缺相关的问题。与基于比对的方法(如HHblits和单特征cnn)不同,所提出的GenEfflux框架将生成序列扩展与通过位置特定评分矩阵(PSSMs)进行多尺度进化特征提取相结合,从而增强了对序列-函数关系的理解。在比较评估中,GenEfflux在所有Efflux转运蛋白类别中始终优于基线deepEfflux模型。B类的敏感性从0.5385上升到0.9999,Matthews相关系数(MCC)从0.4397上升到0.9327。在C类中,准确度从0.8977提高到0.9668,MCC从0.7668提高到0.9331。研究结果表明,ProtGPT2生成的序列具有功能相关性,提高了分类效率。GenEfflux为加强外排蛋白分析和推进抗生素耐药性研究提供了一个全面的框架。
本文章由计算机程序翻译,如有差异,请以英文原文为准。
求助全文
约1分钟内获得全文
求助全文
来源期刊

Computational Biology and Chemistry
生物-计算机:跨学科应用
CiteScore
6.10
自引率
3.20%
发文量
142
审稿时长
24 days
期刊介绍:
Computational Biology and Chemistry publishes original research papers and review articles in all areas of computational life sciences. High quality research contributions with a major computational component in the areas of nucleic acid and protein sequence research, molecular evolution, molecular genetics (functional genomics and proteomics), theory and practice of either biology-specific or chemical-biology-specific modeling, and structural biology of nucleic acids and proteins are particularly welcome. Exceptionally high quality research work in bioinformatics, systems biology, ecology, computational pharmacology, metabolism, biomedical engineering, epidemiology, and statistical genetics will also be considered.
Given their inherent uncertainty, protein modeling and molecular docking studies should be thoroughly validated. In the absence of experimental results for validation, the use of molecular dynamics simulations along with detailed free energy calculations, for example, should be used as complementary techniques to support the major conclusions. Submissions of premature modeling exercises without additional biological insights will not be considered.
Review articles will generally be commissioned by the editors and should not be submitted to the journal without explicit invitation. However prospective authors are welcome to send a brief (one to three pages) synopsis, which will be evaluated by the editors.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


