{"title":"Pontocerebellar Hypoplasia Type 3 With Two Novel <i>PCLO</i> Gene Mutations: A Case Report.","authors":"Sethapong Lertsakulbunlue, Panithi Piyachon, Pitchaya Pichantianchai, Thanapat Chivaruangrot, Piradee Suwanpakdee, Boonchai Boonyawat","doi":"10.1155/crpe/1955363","DOIUrl":null,"url":null,"abstract":"<p><p>Pontocerebellar hypoplasia Type III (PCH3) is a rare, autosomal recessive neurodegenerative disorder linked to mutations in the <i>PCLO</i> gene, previously reported only in Omani populations. It presents with progressive microcephaly, intractable epilepsy, optic atrophy, and severe developmental delay. Here, we report the first documented case of PCH3 in an 8-year-old Thai girl with two novel <i>PCLO</i> truncation mutations. The patient presented with intractable epilepsy from 2 months of age and severe global developmental delay. Whole exome sequencing identified compound heterozygous mutations in the <i>PCLO</i> gene: c.9018_9037del (p.Tyr3007Ter) and c.8456del (p.Ala2819GlufsTer2), both of which were inherited from heterozygous parents. These mutations are predicted to result in a loss of Piccolo protein function. This case expands the mutational spectrum of <i>PCLO</i>-related PCH3 and highlights the importance of advanced molecular diagnostics in understanding and managing this rare neurodegenerative disorder. Given the lack of curative therapies, early genetic diagnosis is crucial in guiding patient care and genetic counseling.</p>","PeriodicalId":9623,"journal":{"name":"Case Reports in Pediatrics","volume":"2025 ","pages":"1955363"},"PeriodicalIF":0.5000,"publicationDate":"2025-07-07","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12259299/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Case Reports in Pediatrics","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1155/crpe/1955363","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2025/1/1 0:00:00","PubModel":"eCollection","JCR":"Q4","JCRName":"PEDIATRICS","Score":null,"Total":0}
引用次数: 0
Abstract
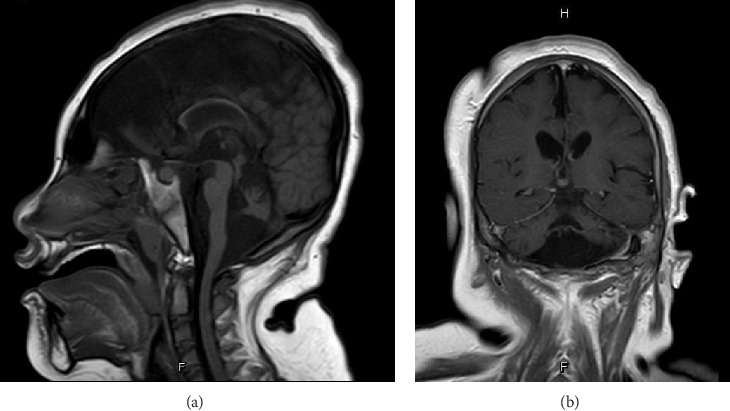
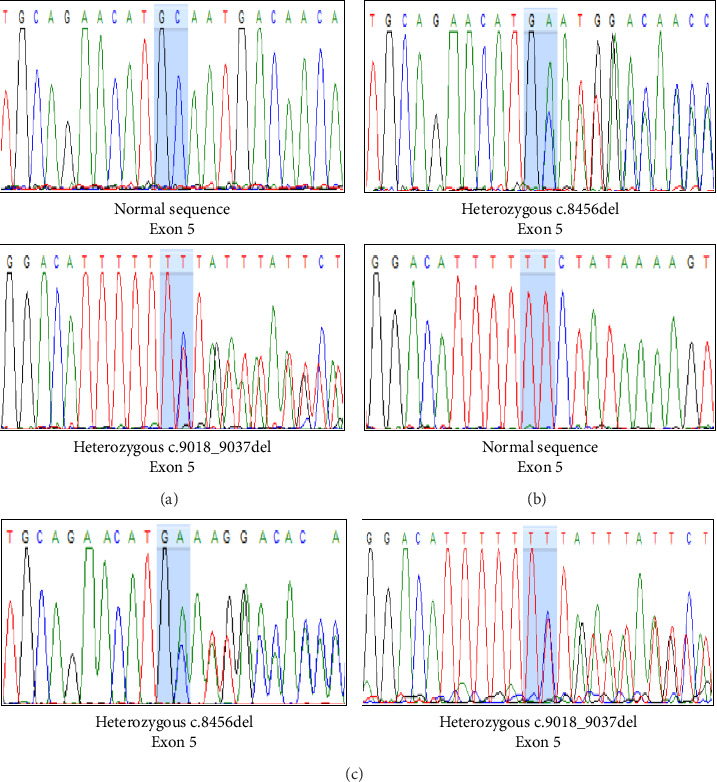
Pontocerebellar hypoplasia Type III (PCH3) is a rare, autosomal recessive neurodegenerative disorder linked to mutations in the PCLO gene, previously reported only in Omani populations. It presents with progressive microcephaly, intractable epilepsy, optic atrophy, and severe developmental delay. Here, we report the first documented case of PCH3 in an 8-year-old Thai girl with two novel PCLO truncation mutations. The patient presented with intractable epilepsy from 2 months of age and severe global developmental delay. Whole exome sequencing identified compound heterozygous mutations in the PCLO gene: c.9018_9037del (p.Tyr3007Ter) and c.8456del (p.Ala2819GlufsTer2), both of which were inherited from heterozygous parents. These mutations are predicted to result in a loss of Piccolo protein function. This case expands the mutational spectrum of PCLO-related PCH3 and highlights the importance of advanced molecular diagnostics in understanding and managing this rare neurodegenerative disorder. Given the lack of curative therapies, early genetic diagnosis is crucial in guiding patient care and genetic counseling.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


