Separation mechanism and chiral-HPLC chromatogram profile of racemic mandelate compounds: a comparative study between experiment and computation using conformer–rotamer ensemble sampling tool (CREST)-XTB
{"title":"Separation mechanism and chiral-HPLC chromatogram profile of racemic mandelate compounds: a comparative study between experiment and computation using conformer–rotamer ensemble sampling tool (CREST)-XTB","authors":"Fenti Fatmawati, Aditya Wibawa Sakti, Suci Zulaikha Hildayani, Akhmaloka, Fida Madayanti Warganegara, Muhamad Abdulkadir Martoprawiro","doi":"10.1007/s00894-025-06408-6","DOIUrl":null,"url":null,"abstract":"<div><h3>Context</h3><p>Nearly 90% of drugs on the market are racemates. A racemate is a mixture of two enantiomers or substances in equal amounts that have an asymmetric molecular structure that is a mirror image of each other. Despite having the same chemical structure, chiral drug isomers can exhibit very different biological behaviors in terms of pharmacology, toxicity, pharmacokinetics, metabolism, etc. Since racemic drugs have only one bioactive enantiomer while its counterpart enantiomers impart undesirable pharmacological properties, it is necessary to separate these racemic compounds to obtain the desired active enantiomer. Chromatography is one of the approaches for the separation of enantiomers. In this study, we observed the chromatographic profile of racemic mandelic acid compound passed through a chiral HPLC column. The chromatogram profile was then observed computationally to study the separation mechanism. The experimental results are in line with the computational analysis that the S chromatogram eluted first compared to the R-enantiomer. It can be predicted that the binding energy of the R-enantiomer (–108.92 kJ/mol) is stronger than the S-enantiomer (− 67 kJ/mol).</p><h3>Methods</h3><p>The chromatogram profile of mandelic acid racemate was observed experimentally using a chiral OD column, and the prediction of column-ligand binding energy was based on computational studies using the conformer–rotamer ensemble sampling tool (CREST). The chromatogram profile was identified using a 0.46 cm × 25 cm chiral OD column HPLC instrument (Daicel Chemical). The samples used were racemic compounds of mandelic acid and standard (S)-mandelic acid. Computational calculations of column capacity factors and binding energies of each enantiomer were performed with a Windows 11 Pro 64-bit operating system, × 64-based processor, equipped with the MGL-Tools program consisting of the ADT (Autodock Tools) application, Avogadro, AutoDock 4.2, Discovery Studio 2020 Client®, and CREST installed as a driver program for the XTB semiempirical quantum chemistry package. For geometry optimization and sampling of DMPC-ligand complexes, we used CREST at https://github.com/grimme-lab/crest.</p></div>","PeriodicalId":651,"journal":{"name":"Journal of Molecular Modeling","volume":"31 8","pages":""},"PeriodicalIF":2.5000,"publicationDate":"2025-07-12","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Molecular Modeling","FirstCategoryId":"92","ListUrlMain":"https://link.springer.com/article/10.1007/s00894-025-06408-6","RegionNum":4,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q4","JCRName":"BIOCHEMISTRY & MOLECULAR BIOLOGY","Score":null,"Total":0}
引用次数: 0
Abstract
Context
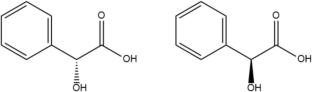
Nearly 90% of drugs on the market are racemates. A racemate is a mixture of two enantiomers or substances in equal amounts that have an asymmetric molecular structure that is a mirror image of each other. Despite having the same chemical structure, chiral drug isomers can exhibit very different biological behaviors in terms of pharmacology, toxicity, pharmacokinetics, metabolism, etc. Since racemic drugs have only one bioactive enantiomer while its counterpart enantiomers impart undesirable pharmacological properties, it is necessary to separate these racemic compounds to obtain the desired active enantiomer. Chromatography is one of the approaches for the separation of enantiomers. In this study, we observed the chromatographic profile of racemic mandelic acid compound passed through a chiral HPLC column. The chromatogram profile was then observed computationally to study the separation mechanism. The experimental results are in line with the computational analysis that the S chromatogram eluted first compared to the R-enantiomer. It can be predicted that the binding energy of the R-enantiomer (–108.92 kJ/mol) is stronger than the S-enantiomer (− 67 kJ/mol).
Methods
The chromatogram profile of mandelic acid racemate was observed experimentally using a chiral OD column, and the prediction of column-ligand binding energy was based on computational studies using the conformer–rotamer ensemble sampling tool (CREST). The chromatogram profile was identified using a 0.46 cm × 25 cm chiral OD column HPLC instrument (Daicel Chemical). The samples used were racemic compounds of mandelic acid and standard (S)-mandelic acid. Computational calculations of column capacity factors and binding energies of each enantiomer were performed with a Windows 11 Pro 64-bit operating system, × 64-based processor, equipped with the MGL-Tools program consisting of the ADT (Autodock Tools) application, Avogadro, AutoDock 4.2, Discovery Studio 2020 Client®, and CREST installed as a driver program for the XTB semiempirical quantum chemistry package. For geometry optimization and sampling of DMPC-ligand complexes, we used CREST at https://github.com/grimme-lab/crest.
期刊介绍:
The Journal of Molecular Modeling focuses on "hardcore" modeling, publishing high-quality research and reports. Founded in 1995 as a purely electronic journal, it has adapted its format to include a full-color print edition, and adjusted its aims and scope fit the fast-changing field of molecular modeling, with a particular focus on three-dimensional modeling.
Today, the journal covers all aspects of molecular modeling including life science modeling; materials modeling; new methods; and computational chemistry.
Topics include computer-aided molecular design; rational drug design, de novo ligand design, receptor modeling and docking; cheminformatics, data analysis, visualization and mining; computational medicinal chemistry; homology modeling; simulation of peptides, DNA and other biopolymers; quantitative structure-activity relationships (QSAR) and ADME-modeling; modeling of biological reaction mechanisms; and combined experimental and computational studies in which calculations play a major role.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


