{"title":"De Novo Splice Site Variant of TCF12 in a Boy With Isolated Kallmann Syndrome.","authors":"Erina Suzuki, Hirohito Shima, Aki Ueda, Kazuhiko Nakabayashi, Keiko Matsubara, Yoko Kuroki, Junko Kanno, Maki Fukami","doi":"10.1155/crie/2350842","DOIUrl":null,"url":null,"abstract":"<p><p><b>Background:</b> Kallmann syndrome is a rare endocrinopathy characterized by congenital hypogonadotropic hypogonadism (CHH) and olfactory dysfunction. The current understanding of the genetic basis of Kallmann syndrome is fragmentary. TCF12 is a causative gene for autosomal dominant craniosynostosis with various complications. Although recent studies identified rare nucleotide substitutions and indels of TCF12 in a few families with CHH and additional clinical features, the significance of TCF12 variants as the cause of Kallmann syndrome remains uncertain. <b>Case Presentation:</b> A Japanese boy exhibited bilateral cryptorchidism and micropenis at birth. He was otherwise healthy and had normal developmental milestones. At 11 years of age, he showed no pubertal signs. Physical examinations detected no clinical abnormalities except hyposmia. Brain imaging suggested olfactory bulb hypoplasia, but no other anomalies. A gonadotropin-releasing hormone (GnRH) stimulation test yielded low responses of gonadotropins. Whole-exome sequencing (WES) identified a hitherto unreported de novo heterozygous substitution at a splice acceptor site of TCF12 (c.391-1G >A). This variant was predicted to cause a frameshift and resultant premature termination (p.Ser132ProfsTer38) and was assessed as pathogenic, according to the ACMG/AMP 2015 guidelines. The patient carried no rare variants in other genes previously associated with Kallmann syndrome or CHH. <b>Conclusion:</b> These results broaden the mutation spectrum of TCF12. More importantly, this study argues for the etiological relationship between TCF12 variants and isolated Kallmann syndrome.</p>","PeriodicalId":9621,"journal":{"name":"Case Reports in Endocrinology","volume":"2025 ","pages":"2350842"},"PeriodicalIF":0.9000,"publicationDate":"2025-07-03","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12245484/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Case Reports in Endocrinology","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1155/crie/2350842","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2025/1/1 0:00:00","PubModel":"eCollection","JCR":"Q4","JCRName":"ENDOCRINOLOGY & METABOLISM","Score":null,"Total":0}
引用次数: 0
Abstract
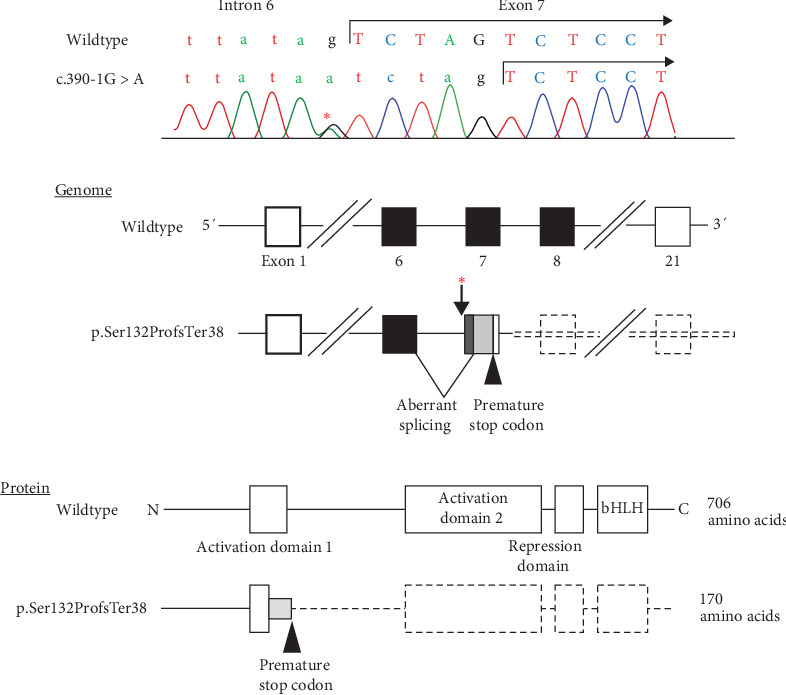
Background: Kallmann syndrome is a rare endocrinopathy characterized by congenital hypogonadotropic hypogonadism (CHH) and olfactory dysfunction. The current understanding of the genetic basis of Kallmann syndrome is fragmentary. TCF12 is a causative gene for autosomal dominant craniosynostosis with various complications. Although recent studies identified rare nucleotide substitutions and indels of TCF12 in a few families with CHH and additional clinical features, the significance of TCF12 variants as the cause of Kallmann syndrome remains uncertain. Case Presentation: A Japanese boy exhibited bilateral cryptorchidism and micropenis at birth. He was otherwise healthy and had normal developmental milestones. At 11 years of age, he showed no pubertal signs. Physical examinations detected no clinical abnormalities except hyposmia. Brain imaging suggested olfactory bulb hypoplasia, but no other anomalies. A gonadotropin-releasing hormone (GnRH) stimulation test yielded low responses of gonadotropins. Whole-exome sequencing (WES) identified a hitherto unreported de novo heterozygous substitution at a splice acceptor site of TCF12 (c.391-1G >A). This variant was predicted to cause a frameshift and resultant premature termination (p.Ser132ProfsTer38) and was assessed as pathogenic, according to the ACMG/AMP 2015 guidelines. The patient carried no rare variants in other genes previously associated with Kallmann syndrome or CHH. Conclusion: These results broaden the mutation spectrum of TCF12. More importantly, this study argues for the etiological relationship between TCF12 variants and isolated Kallmann syndrome.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


