Sang Heon Suh, Su Hyun Song, Hong Sang Choi, Chang Seong Kim, Eun Hui Bae, Soo Wan Kim, Seong Kwon Ma
{"title":"A Case of Cystinuria With Compound Heterozygous Mutations Both in <i>SLC3A1</i> and <i>SLC7A9</i> Genes.","authors":"Sang Heon Suh, Su Hyun Song, Hong Sang Choi, Chang Seong Kim, Eun Hui Bae, Soo Wan Kim, Seong Kwon Ma","doi":"10.5049/EBP.2025.23.e2","DOIUrl":null,"url":null,"abstract":"<p><p>Cystinuria is an autosomal recessively inherited genetic disorder, and is typically classified into type A, caused by mutations in <i>SLC3A1</i>, or type B, caused by mutations in <i>SLC7A9</i>. While the predominance of the genotypes varies among countries, due to lack of a large scale cohort, the characterization of mutations in <i>SLC3A1</i> or <i>SLC7A9</i> is still limited in East Asia. A 61-year-old male patient admitted to the department of nephrology, with a chief complaint of fever, chillness and left flank pain for a week. The patient had a past history of recurrent urolithiasis, with a frequency of at least 1 to 2 times a year. Computed tomography visualized 1 cm-sized stone at distal ureter, which was removed by retrograde ureteroscopy. The stone analysis documented 100% of cystine, indicating an underlying genetic disorder, cystinuria. Whole genome sequencing from peripheral blood unveiled 3 heterozygous missense mutations in coding exons of <i>SLC3A1</i> gene, and 2 heterozygous missense mutations in coding exons of <i>SLC7A9</i> gene. We here report a case of cystinuria with compound heterozygous mutations both in <i>SLC3A1</i> and <i>SLC7A9</i> genes, with a total of 5 mutant alleles in a patient.</p>","PeriodicalId":35352,"journal":{"name":"Electrolyte and Blood Pressure","volume":"23 1","pages":"17-21"},"PeriodicalIF":0.0000,"publicationDate":"2025-06-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12230274/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Electrolyte and Blood Pressure","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.5049/EBP.2025.23.e2","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2025/6/23 0:00:00","PubModel":"Epub","JCR":"Q3","JCRName":"Medicine","Score":null,"Total":0}
引用次数: 0
Abstract
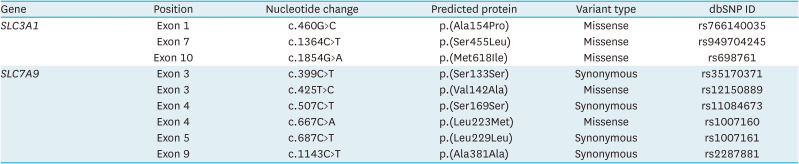
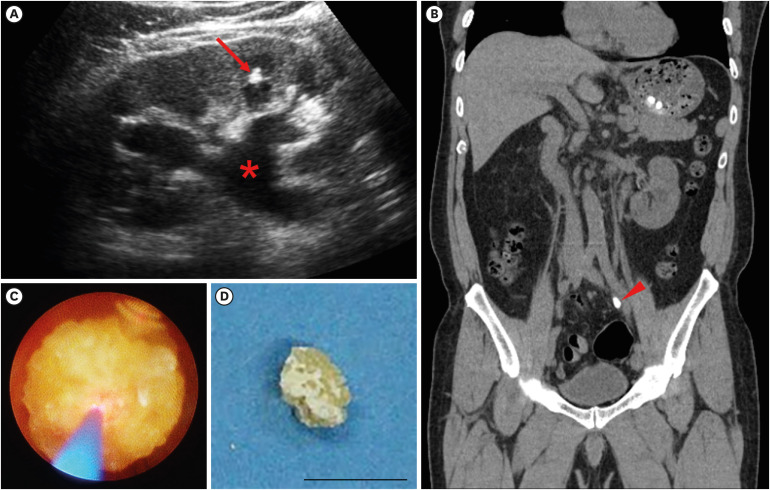
Cystinuria is an autosomal recessively inherited genetic disorder, and is typically classified into type A, caused by mutations in SLC3A1, or type B, caused by mutations in SLC7A9. While the predominance of the genotypes varies among countries, due to lack of a large scale cohort, the characterization of mutations in SLC3A1 or SLC7A9 is still limited in East Asia. A 61-year-old male patient admitted to the department of nephrology, with a chief complaint of fever, chillness and left flank pain for a week. The patient had a past history of recurrent urolithiasis, with a frequency of at least 1 to 2 times a year. Computed tomography visualized 1 cm-sized stone at distal ureter, which was removed by retrograde ureteroscopy. The stone analysis documented 100% of cystine, indicating an underlying genetic disorder, cystinuria. Whole genome sequencing from peripheral blood unveiled 3 heterozygous missense mutations in coding exons of SLC3A1 gene, and 2 heterozygous missense mutations in coding exons of SLC7A9 gene. We here report a case of cystinuria with compound heterozygous mutations both in SLC3A1 and SLC7A9 genes, with a total of 5 mutant alleles in a patient.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


