{"title":"Novel Loss of Function Variant in SOST From Chinese Family Results in Sclerosteosis 1.","authors":"Yufan Guo, Xintao Wu, Yuting Jin, Yu Gu, Yuting Lou, Pu Miao, Ye Wang, Bijun Zhang, Xueting Lin, Chudi Zhang, Jianhua Feng","doi":"10.1002/mgg3.70109","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>SOST encodes a secreted glycoprotein that is similar in sequence to the differential screening-selected gene aberrative in neuroblastoma (DAN) family of bone morphogenetic protein (BMP) antagonists. Pathogenic variants in the SOST gene result in sclerosteosis, van Buchem disease (VBD), or craniodiaphyseal dysplasia. SOST-related genetic disorders are very rare, and limited studies have reported variants associated with sclerosteosis.</p><p><strong>Methods: </strong>Clinical tests such as magnetic resonance imaging (MRI), computed tomography (CT), emission computed tomography (ECT), electromyogram (EMG), routine blood tests, and physical examinations were conducted for the proband. Trio-whole exome sequencing (Trio-WES) was performed, and the rare variants (allele frequency < 0.01) in the exon and splicing regions were selected for further pathogenic evaluation. Candidate pathogenic variants were validated through Sanger sequencing. The wild and mutant SOST sequences were cloned into the pcDNA3.1 expression vector, and the RNA and protein expression levels were investigated in the HEK293T cell line.</p><p><strong>Results: </strong>In this study, we present a case study of a proband who displays abnormal facial expressions accompanied by numbness. The results of the brain MRI show thickening of the skull and disappearance of the diplopia signal. The temporal bone CT scan indicates diffuse osteosclerosis affecting the bilateral ossicular chains and internal auditory meatus, as well as stenosis of the bilateral internal auditory meatus. Trio-WES sequencing detected a novel homozygous variant in the proband: NM_025237.3(SOST): c.327C>A (p.Cys109*), which was also validated in his sister from the same family. According to the ACMG guidelines, the variant is classified as \"likely pathogenic.\" The in vitro experiments demonstrated that the variant caused a decrease in SOST expression at RNA and protein level and produced a truncated protein.</p><p><strong>Conclusion: </strong>The report presents new evidence for the clinical diagnosis of SOST-related facial numbness and expands the variant spectrum of SOST.</p>","PeriodicalId":18852,"journal":{"name":"Molecular Genetics & Genomic Medicine","volume":"13 7","pages":"e70109"},"PeriodicalIF":1.6000,"publicationDate":"2025-07-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12222177/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Molecular Genetics & Genomic Medicine","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1002/mgg3.70109","RegionNum":4,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q4","JCRName":"GENETICS & HEREDITY","Score":null,"Total":0}
引用次数: 0
Abstract
Background: SOST encodes a secreted glycoprotein that is similar in sequence to the differential screening-selected gene aberrative in neuroblastoma (DAN) family of bone morphogenetic protein (BMP) antagonists. Pathogenic variants in the SOST gene result in sclerosteosis, van Buchem disease (VBD), or craniodiaphyseal dysplasia. SOST-related genetic disorders are very rare, and limited studies have reported variants associated with sclerosteosis.
Methods: Clinical tests such as magnetic resonance imaging (MRI), computed tomography (CT), emission computed tomography (ECT), electromyogram (EMG), routine blood tests, and physical examinations were conducted for the proband. Trio-whole exome sequencing (Trio-WES) was performed, and the rare variants (allele frequency < 0.01) in the exon and splicing regions were selected for further pathogenic evaluation. Candidate pathogenic variants were validated through Sanger sequencing. The wild and mutant SOST sequences were cloned into the pcDNA3.1 expression vector, and the RNA and protein expression levels were investigated in the HEK293T cell line.
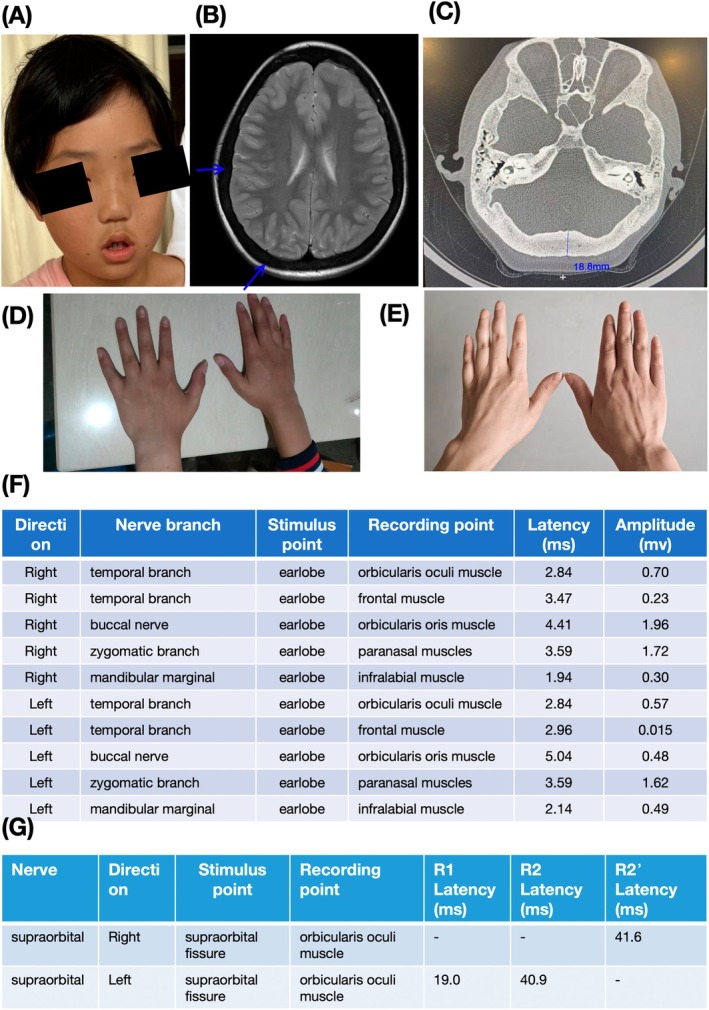
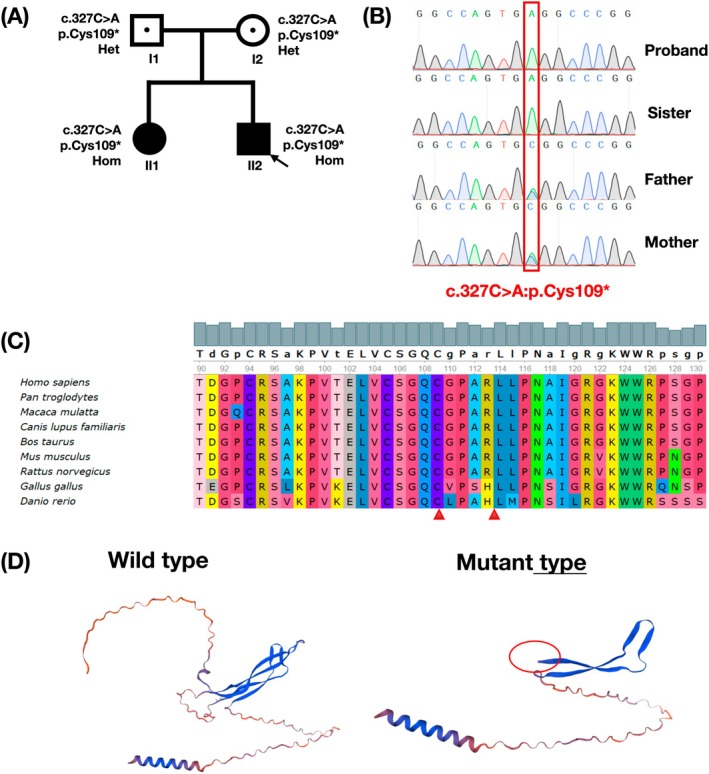
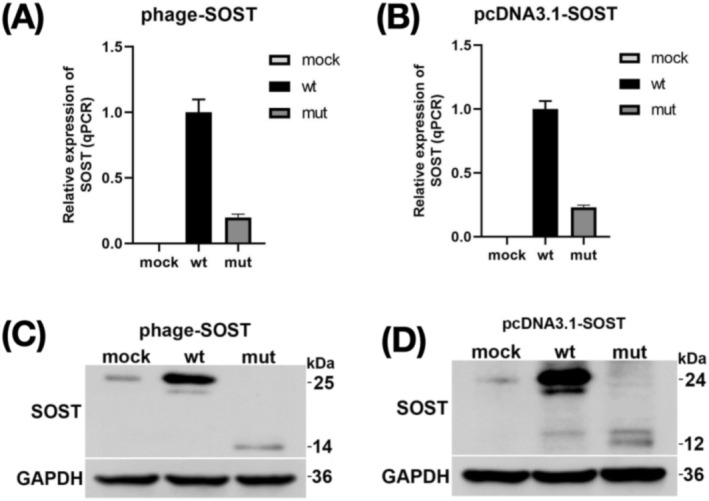
Results: In this study, we present a case study of a proband who displays abnormal facial expressions accompanied by numbness. The results of the brain MRI show thickening of the skull and disappearance of the diplopia signal. The temporal bone CT scan indicates diffuse osteosclerosis affecting the bilateral ossicular chains and internal auditory meatus, as well as stenosis of the bilateral internal auditory meatus. Trio-WES sequencing detected a novel homozygous variant in the proband: NM_025237.3(SOST): c.327C>A (p.Cys109*), which was also validated in his sister from the same family. According to the ACMG guidelines, the variant is classified as "likely pathogenic." The in vitro experiments demonstrated that the variant caused a decrease in SOST expression at RNA and protein level and produced a truncated protein.
Conclusion: The report presents new evidence for the clinical diagnosis of SOST-related facial numbness and expands the variant spectrum of SOST.
背景:SOST编码一种分泌的糖蛋白,其序列与成神经细胞瘤(DAN)骨形态发生蛋白(BMP)拮抗剂家族的差异筛选基因畸变相似。SOST基因的致病变异导致硬化、van Buchem病(VBD)或颅干发育不良。sost相关的遗传疾病非常罕见,有限的研究报告了与硬化症相关的变异。方法:对先证者进行磁共振成像(MRI)、计算机断层扫描(CT)、发射计算机断层扫描(ECT)、肌电图(EMG)、血常规、体格检查等临床检查。三全外显子组测序(Trio-WES)进行,罕见变异(等位基因频率)结果:在本研究中,我们提出了一个先证者的病例研究,他表现出异常的面部表情并伴有麻木。脑部MRI结果显示颅骨增厚,复视信号消失。颞骨CT示弥漫性骨硬化累及双侧听骨链及内耳道,双侧内耳道狭窄。Trio-WES测序在先证者NM_025237.3(SOST): c.327C> a (p.Cys109*)中检测到一个新的纯合变异,该变异在同一家族的姐妹中也得到了验证。根据ACMG指南,这种变异被归类为“可能致病”。体外实验表明,该变异在RNA和蛋白水平上导致SOST表达降低,并产生一个截断的蛋白。结论:本报告为SOST相关面部麻木的临床诊断提供了新的依据,扩大了SOST的变异谱。
期刊介绍:
Molecular Genetics & Genomic Medicine is a peer-reviewed journal for rapid dissemination of quality research related to the dynamically developing areas of human, molecular and medical genetics. The journal publishes original research articles covering findings in phenotypic, molecular, biological, and genomic aspects of genomic variation, inherited disorders and birth defects. The broad publishing spectrum of Molecular Genetics & Genomic Medicine includes rare and common disorders from diagnosis to treatment. Examples of appropriate articles include reports of novel disease genes, functional studies of genetic variants, in-depth genotype-phenotype studies, genomic analysis of inherited disorders, molecular diagnostic methods, medical bioinformatics, ethical, legal, and social implications (ELSI), and approaches to clinical diagnosis. Molecular Genetics & Genomic Medicine provides a scientific home for next generation sequencing studies of rare and common disorders, which will make research in this fascinating area easily and rapidly accessible to the scientific community. This will serve as the basis for translating next generation sequencing studies into individualized diagnostics and therapeutics, for day-to-day medical care.
Molecular Genetics & Genomic Medicine publishes original research articles, reviews, and research methods papers, along with invited editorials and commentaries. Original research papers must report well-conducted research with conclusions supported by the data presented.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


