{"title":"A DFT study of the hydrolytic degradation mechanisms of iprovalicarb and iprodione: implications in environmental safety","authors":"Peter N. Nelson","doi":"10.1007/s00894-025-06424-6","DOIUrl":null,"url":null,"abstract":"<div><h3>Context</h3><p>Iprovalicarb and iprodione represent two popular carboxylic acid amide-type (CAA) fungicides which have found wide spread application in the protection of various crops. However, though useful, excessive usage of these compounds could have deleterious effects on human health and environmental safety. Hence, a thorough DFT investigation of the degradation thermodynamics and mechanism for these two compounds was carried out, revealing a multi-step hydrolytic transformation process in the gas phase via overall exergonic processes where the rate limiting steps are calculated at ca. 197 and 235 kJ mol<sup>−1</sup>, for iprodione and iprovalicarb, respectively. However, in aqueous media, whereas for iprodione the hydrolysis mechanism is identical to that in the gas phase, for iprovalicarb, the endergonic solution phase mechanism is different. Overall, both compounds undergo very slow hydrolysis at neutral pH but, of the two, iprodione offers the shortest residence time. </p><h3>Methods</h3><p>All calculations were carried out at the 6–311 + + G(d,p)/CAM-B3LYP level of theory, as implemented in the Gaussian-16 software suite. Solution phase calculations were carried out via the well-regarded C-PCM model, an implicit solvation model, known to be efficient and effective at predicting solvation effects.</p></div>","PeriodicalId":651,"journal":{"name":"Journal of Molecular Modeling","volume":"31 7","pages":""},"PeriodicalIF":2.5000,"publicationDate":"2025-06-28","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Molecular Modeling","FirstCategoryId":"92","ListUrlMain":"https://link.springer.com/article/10.1007/s00894-025-06424-6","RegionNum":4,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q4","JCRName":"BIOCHEMISTRY & MOLECULAR BIOLOGY","Score":null,"Total":0}
引用次数: 0
Abstract
Context
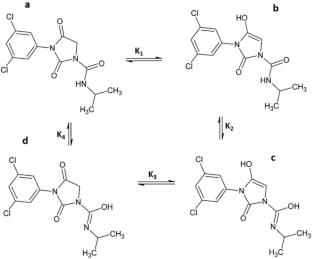
Iprovalicarb and iprodione represent two popular carboxylic acid amide-type (CAA) fungicides which have found wide spread application in the protection of various crops. However, though useful, excessive usage of these compounds could have deleterious effects on human health and environmental safety. Hence, a thorough DFT investigation of the degradation thermodynamics and mechanism for these two compounds was carried out, revealing a multi-step hydrolytic transformation process in the gas phase via overall exergonic processes where the rate limiting steps are calculated at ca. 197 and 235 kJ mol−1, for iprodione and iprovalicarb, respectively. However, in aqueous media, whereas for iprodione the hydrolysis mechanism is identical to that in the gas phase, for iprovalicarb, the endergonic solution phase mechanism is different. Overall, both compounds undergo very slow hydrolysis at neutral pH but, of the two, iprodione offers the shortest residence time.
Methods
All calculations were carried out at the 6–311 + + G(d,p)/CAM-B3LYP level of theory, as implemented in the Gaussian-16 software suite. Solution phase calculations were carried out via the well-regarded C-PCM model, an implicit solvation model, known to be efficient and effective at predicting solvation effects.
期刊介绍:
The Journal of Molecular Modeling focuses on "hardcore" modeling, publishing high-quality research and reports. Founded in 1995 as a purely electronic journal, it has adapted its format to include a full-color print edition, and adjusted its aims and scope fit the fast-changing field of molecular modeling, with a particular focus on three-dimensional modeling.
Today, the journal covers all aspects of molecular modeling including life science modeling; materials modeling; new methods; and computational chemistry.
Topics include computer-aided molecular design; rational drug design, de novo ligand design, receptor modeling and docking; cheminformatics, data analysis, visualization and mining; computational medicinal chemistry; homology modeling; simulation of peptides, DNA and other biopolymers; quantitative structure-activity relationships (QSAR) and ADME-modeling; modeling of biological reaction mechanisms; and combined experimental and computational studies in which calculations play a major role.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


