Extremely High RAHB Energies Quantified via a Molecular Tailoring Approach in Fused-Ring Tropolones and Their Effect on the Electronic Structure and Optical Properties
Andrei V. Afonin, Danuta Rusinska-Roszak, Alexander V. Vashchenko
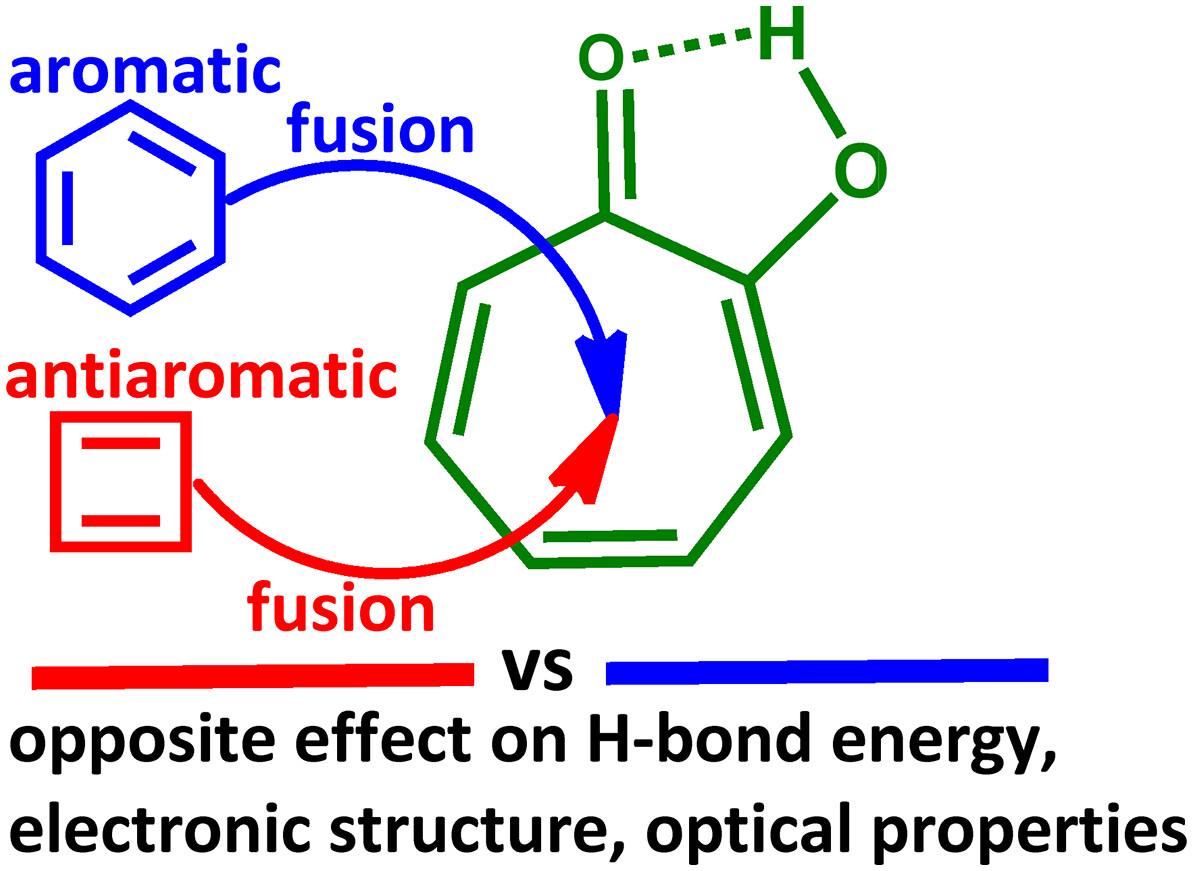
{"title":"Extremely High RAHB Energies Quantified via a Molecular Tailoring Approach in Fused-Ring Tropolones and Their Effect on the Electronic Structure and Optical Properties","authors":"Andrei V. Afonin, Danuta Rusinska-Roszak, Alexander V. Vashchenko","doi":"10.1002/jcc.70153","DOIUrl":null,"url":null,"abstract":"<div>\n \n <p>The O<span></span>H···OC RAHB energies were quantified via a molecular tailoring approach for a series of tropolones with the fused aromatic and antiaromatic cycles. At 3,4 and 5,6 fusions of the tropone ring with aromatic cycles, the RAHB enhancement is observed while the RAHB weakening takes place at 4,5 and 6,7 fusions. An opposite trend appears at the fusion of the tropone ring with the antiaromatic cycle. A linear ratio was found between the HOMA structure-based aromaticity indices as well as NICS magnetic ones for the tropone ring and the RAHB energy. The total RAHB energy is divided into σ- and π-components. The σ- and π-components were established to change in the same directions, appearing the synergism. This synergism causes extremely high RAHB energies above 30 kcal/mol in some tropolones. It was shown that RAHB acquires the features of CAHB in these cases due to the excess charge on the oxygen of the CO group. The influence of the RAHB strength on the frontier molecular orbitals patterns, energy, the size of the gap between them, and the <i>λ</i><sub>max</sub> absorption wavelength in the UV/vis range was studied. It was revealed that the HOMO energy elevates, the LUMO energy lowers, the HOMO-LUMO gap narrows, and the <i>λ</i><sub>max</sub> wavelength undergoes a bathochromic shift with RAHB strengthening in the studied tropolones.</p>\n </div>","PeriodicalId":188,"journal":{"name":"Journal of Computational Chemistry","volume":"46 17","pages":""},"PeriodicalIF":4.8000,"publicationDate":"2025-06-29","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Computational Chemistry","FirstCategoryId":"92","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1002/jcc.70153","RegionNum":3,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"CHEMISTRY, MULTIDISCIPLINARY","Score":null,"Total":0}
引用次数: 0
Abstract
The OH···OC RAHB energies were quantified via a molecular tailoring approach for a series of tropolones with the fused aromatic and antiaromatic cycles. At 3,4 and 5,6 fusions of the tropone ring with aromatic cycles, the RAHB enhancement is observed while the RAHB weakening takes place at 4,5 and 6,7 fusions. An opposite trend appears at the fusion of the tropone ring with the antiaromatic cycle. A linear ratio was found between the HOMA structure-based aromaticity indices as well as NICS magnetic ones for the tropone ring and the RAHB energy. The total RAHB energy is divided into σ- and π-components. The σ- and π-components were established to change in the same directions, appearing the synergism. This synergism causes extremely high RAHB energies above 30 kcal/mol in some tropolones. It was shown that RAHB acquires the features of CAHB in these cases due to the excess charge on the oxygen of the CO group. The influence of the RAHB strength on the frontier molecular orbitals patterns, energy, the size of the gap between them, and the λmax absorption wavelength in the UV/vis range was studied. It was revealed that the HOMO energy elevates, the LUMO energy lowers, the HOMO-LUMO gap narrows, and the λmax wavelength undergoes a bathochromic shift with RAHB strengthening in the studied tropolones.
期刊介绍:
This distinguished journal publishes articles concerned with all aspects of computational chemistry: analytical, biological, inorganic, organic, physical, and materials. The Journal of Computational Chemistry presents original research, contemporary developments in theory and methodology, and state-of-the-art applications. Computational areas that are featured in the journal include ab initio and semiempirical quantum mechanics, density functional theory, molecular mechanics, molecular dynamics, statistical mechanics, cheminformatics, biomolecular structure prediction, molecular design, and bioinformatics.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


