Yaxin Zhang, Qiqin Wu, Ying Zhou, Qingyu Cheng, Tengchuan Jin
{"title":"hDNApipe: streamlining human genome analysis and interpretation with an intuitive and user-friendly interface.","authors":"Yaxin Zhang, Qiqin Wu, Ying Zhou, Qingyu Cheng, Tengchuan Jin","doi":"10.1093/nargab/lqaf088","DOIUrl":null,"url":null,"abstract":"<p><p>With the rapid evolution of next-generation sequencing technology, numerous tools have emerged across multiple stages in the human genome analysis, complicating the assembly of an appropriate pipeline. To address this challenge, there is a pressing need for an efficient and user-friendly tool that combines extensive features with intuitive operation to streamline the process. Here we introduced hDNApipe, a highly flexible end-to-end pipeline tool designed for the analysis and interpretation of human genomic sequencing data. It is developed using bash scripts and the Python standard graphical user interface library Tkinter, which endows it with excellent usability and accessibility. This pipeline directly obtains variants and associated information, and also optionally enables the visualization of variants and downstream analysis. hDNApipe features dual-mode operation with both the command-line interface and graphical user interface, and provides multiple parameter options that enable users to conduct customized analysis. It features an extraordinarily convenient installation process with a dedicated docker setup, eliminating the complexity of manually installing dependencies. It has been tested on a Linux server using publicly available data. Furthermore, benchmarking with other available pipelines was conducted from alignment to variant calling, demonstrating hDNApipe's outstanding performance in terms of time consumption, precision, and sensitivity.</p>","PeriodicalId":33994,"journal":{"name":"NAR Genomics and Bioinformatics","volume":"7 2","pages":"lqaf088"},"PeriodicalIF":2.8000,"publicationDate":"2025-06-26","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12199140/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"NAR Genomics and Bioinformatics","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1093/nargab/lqaf088","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2025/6/1 0:00:00","PubModel":"eCollection","JCR":"Q1","JCRName":"GENETICS & HEREDITY","Score":null,"Total":0}
引用次数: 0
Abstract
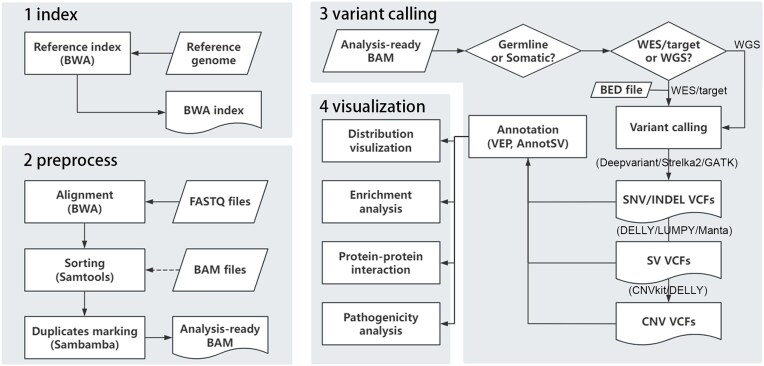
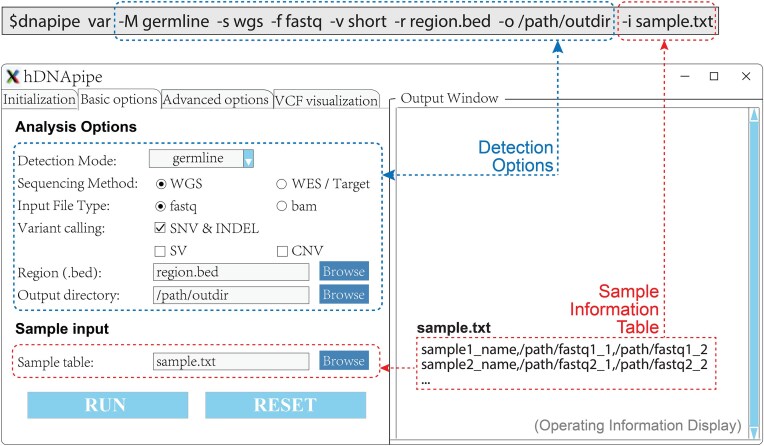
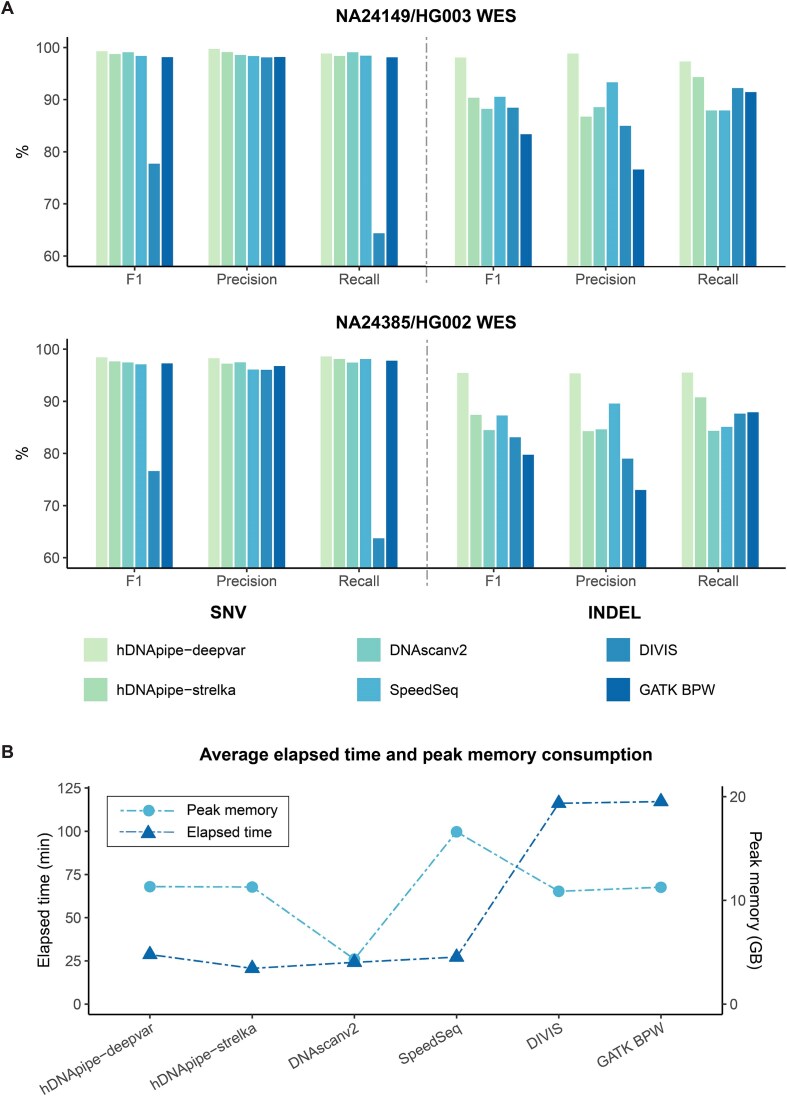
With the rapid evolution of next-generation sequencing technology, numerous tools have emerged across multiple stages in the human genome analysis, complicating the assembly of an appropriate pipeline. To address this challenge, there is a pressing need for an efficient and user-friendly tool that combines extensive features with intuitive operation to streamline the process. Here we introduced hDNApipe, a highly flexible end-to-end pipeline tool designed for the analysis and interpretation of human genomic sequencing data. It is developed using bash scripts and the Python standard graphical user interface library Tkinter, which endows it with excellent usability and accessibility. This pipeline directly obtains variants and associated information, and also optionally enables the visualization of variants and downstream analysis. hDNApipe features dual-mode operation with both the command-line interface and graphical user interface, and provides multiple parameter options that enable users to conduct customized analysis. It features an extraordinarily convenient installation process with a dedicated docker setup, eliminating the complexity of manually installing dependencies. It has been tested on a Linux server using publicly available data. Furthermore, benchmarking with other available pipelines was conducted from alignment to variant calling, demonstrating hDNApipe's outstanding performance in terms of time consumption, precision, and sensitivity.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


