{"title":"Modern methods in peach (Prunus persica) genome research.","authors":"I V Rozanova, E A Vodiasova","doi":"10.18699/vjgb-25-39","DOIUrl":null,"url":null,"abstract":"<p><p>Peach (Prunus persica (L.) Batsch) is one of the main agricultural stone fruit crops of the family Rosaceae. Modern breeding is aimed at improving the quality of the fruit, extending the period of its production, increasing its resistance to unfavorable environmental conditions and reducing the total cost of production of cultivated varieties. However, peach breeding is an extremely long process: it takes 10-15 years from hybridization of the parental forms to obtaining fruit-bearing trees. Research into peach varieties as donors of desirable traits began in the 1980s. The first version of the peach genome was presented in 2013, and its appearance contributed to the identification and localization of loci, followed by the identification of candidate genes that control the desired trait. The development of NGS has accelerated the development of methods based on the use of diagnostic DNA markers. Approaches that allow accelerating classical breeding processes include marker-oriented selection (MOS) and genomic selection. In order to develop DNA markers associated with the traits under investigation, it is necessary to carry out preliminary mapping of loci controlling economically desirable traits and to develop linkage maps. SNP-chip approaches and genotyping by sequencing (GBS) methods are being developed. In recent years, genome-wide association analysis (GWAS) has been actively used to identify genomic loci associated with economically important traits, which requires screening of large samples of varieties for hundreds and thousands of SNPs. Study on the pangenome has shown the need to analyze a larger number of samples, since there is still not enough data to identify polymorphic regions of the genome. The aim of this review was to systematize and summarize the major advances in peach genomic research over the last 40 years: linkage and physical map construction, development of different molecular markers, full genome sequencing for peach, and existing methods for genome-wide association studies with high-density SNP markers. This review provides a theoretical basis for future GWAS analysis in order to identify high-performance markers of economically valuable traits for peach and to develop genomic selection of this crop.</p>","PeriodicalId":44339,"journal":{"name":"Vavilovskii Zhurnal Genetiki i Selektsii","volume":"29 3","pages":"358-369"},"PeriodicalIF":1.0000,"publicationDate":"2025-06-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12188001/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Vavilovskii Zhurnal Genetiki i Selektsii","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.18699/vjgb-25-39","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"AGRICULTURE, MULTIDISCIPLINARY","Score":null,"Total":0}
引用次数: 0
Abstract
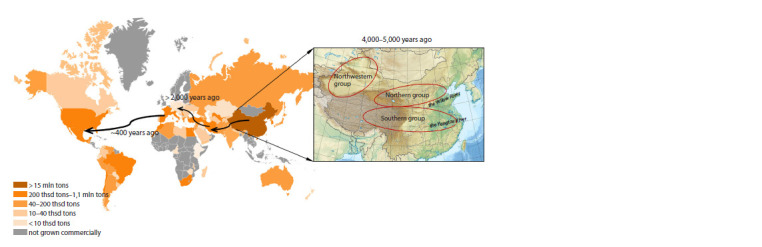
Peach (Prunus persica (L.) Batsch) is one of the main agricultural stone fruit crops of the family Rosaceae. Modern breeding is aimed at improving the quality of the fruit, extending the period of its production, increasing its resistance to unfavorable environmental conditions and reducing the total cost of production of cultivated varieties. However, peach breeding is an extremely long process: it takes 10-15 years from hybridization of the parental forms to obtaining fruit-bearing trees. Research into peach varieties as donors of desirable traits began in the 1980s. The first version of the peach genome was presented in 2013, and its appearance contributed to the identification and localization of loci, followed by the identification of candidate genes that control the desired trait. The development of NGS has accelerated the development of methods based on the use of diagnostic DNA markers. Approaches that allow accelerating classical breeding processes include marker-oriented selection (MOS) and genomic selection. In order to develop DNA markers associated with the traits under investigation, it is necessary to carry out preliminary mapping of loci controlling economically desirable traits and to develop linkage maps. SNP-chip approaches and genotyping by sequencing (GBS) methods are being developed. In recent years, genome-wide association analysis (GWAS) has been actively used to identify genomic loci associated with economically important traits, which requires screening of large samples of varieties for hundreds and thousands of SNPs. Study on the pangenome has shown the need to analyze a larger number of samples, since there is still not enough data to identify polymorphic regions of the genome. The aim of this review was to systematize and summarize the major advances in peach genomic research over the last 40 years: linkage and physical map construction, development of different molecular markers, full genome sequencing for peach, and existing methods for genome-wide association studies with high-density SNP markers. This review provides a theoretical basis for future GWAS analysis in order to identify high-performance markers of economically valuable traits for peach and to develop genomic selection of this crop.
期刊介绍:
The "Vavilov Journal of genetics and breeding" publishes original research and review articles in all key areas of modern plant, animal and human genetics, genomics, bioinformatics and biotechnology. One of the main objectives of the journal is integration of theoretical and applied research in the field of genetics. Special attention is paid to the most topical areas in modern genetics dealing with global concerns such as food security and human health.


 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


