Merve Ayyildiz, Jakob Noske, Florian J. Gisdon, Josef P. Kynast, Birte Höcker
{"title":"Evaluation of Physics-Based Protein Design Methods for Predicting Single Residue Effects on Peptide Binding Specificities","authors":"Merve Ayyildiz, Jakob Noske, Florian J. Gisdon, Josef P. Kynast, Birte Höcker","doi":"10.1002/jcc.70160","DOIUrl":null,"url":null,"abstract":"<p>Understanding the interactions that make up protein–protein or protein-peptide interfaces is a crucial step towards applications in biotechnology. The mutation of a single residue can have a strong impact on binding affinity and specificity, which is difficult to capture in sampling and scoring. Many established computational methods provide an estimate of binding or non-binding; however, comparing highly similar ligands is an important and significantly more challenging problem. Here we evaluated the capability of predicting ligand binding specificity using three established but conceptually different physics-based methods for protein design. As a model system, we analyzed the binding of peptides to designed armadillo repeat proteins, where a single residue of the peptide was changed systematically, leading to affinity changes in the range of 1–1000 nM. We critically assessed the prediction accuracy of the computational methods. While a good correlation with experimentally determined data was observed in several cases, specific biases in the prediction performance of each method were identified.</p>","PeriodicalId":188,"journal":{"name":"Journal of Computational Chemistry","volume":"46 17","pages":""},"PeriodicalIF":4.8000,"publicationDate":"2025-06-26","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://onlinelibrary.wiley.com/doi/epdf/10.1002/jcc.70160","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Computational Chemistry","FirstCategoryId":"92","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1002/jcc.70160","RegionNum":3,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"CHEMISTRY, MULTIDISCIPLINARY","Score":null,"Total":0}
引用次数: 0
Abstract
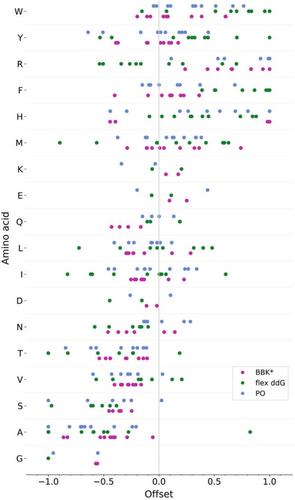
Understanding the interactions that make up protein–protein or protein-peptide interfaces is a crucial step towards applications in biotechnology. The mutation of a single residue can have a strong impact on binding affinity and specificity, which is difficult to capture in sampling and scoring. Many established computational methods provide an estimate of binding or non-binding; however, comparing highly similar ligands is an important and significantly more challenging problem. Here we evaluated the capability of predicting ligand binding specificity using three established but conceptually different physics-based methods for protein design. As a model system, we analyzed the binding of peptides to designed armadillo repeat proteins, where a single residue of the peptide was changed systematically, leading to affinity changes in the range of 1–1000 nM. We critically assessed the prediction accuracy of the computational methods. While a good correlation with experimentally determined data was observed in several cases, specific biases in the prediction performance of each method were identified.
期刊介绍:
This distinguished journal publishes articles concerned with all aspects of computational chemistry: analytical, biological, inorganic, organic, physical, and materials. The Journal of Computational Chemistry presents original research, contemporary developments in theory and methodology, and state-of-the-art applications. Computational areas that are featured in the journal include ab initio and semiempirical quantum mechanics, density functional theory, molecular mechanics, molecular dynamics, statistical mechanics, cheminformatics, biomolecular structure prediction, molecular design, and bioinformatics.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


